靶向治疗时代,如何确定临床药物剂量?——以BTKi、PI3Ki为例
时间:2022-10-13 16:03:03 热度:37.1℃ 作者:网络
针对PI3K、BTK和BCL-2的靶向治疗,已被批准用于慢性淋巴细胞白血病(CLL)。这些疗法作用于非常明确的靶点,但在I期试验中确定其II期剂量时,仍只能从有限的方面进行考虑。
例如,在各种BTK抑制剂的剂量探索研究中,虽已对不可逆抑制剂对BTK激酶的占有率进行了评估,但尚未确定导致BTK激酶完全被占用的最低剂量[1]。
靶向药物与细胞毒药物有不同的剂量反应关系(dose-response relationship)。细胞毒药物在达到最大的肿瘤抑制作用前已达到最大耐受剂量,在最大耐受剂量之前剂量-效应曲线的斜率均较大,因此,临床上应采用其最大安全剂量。而分子靶向治疗药物的毒性较轻,往往在达到最大耐受剂量前已经达到靶点饱和,发挥了最大的肿瘤抑制作用。此时,如果继续增加剂量,疗效无明显增强,反而增加不必要的毒性。
因此,靶向治疗药物的应用剂量应是最佳生物学剂量(optimal biological dose,OBD)[2]。但传统的3 + 3毒性驱动试验设计,仍在靶向治疗时代大量使用。如果更严格地使用药效学生物标志物来指导剂量选择,推荐的II期剂量可能会低于毒性驱动的选择。减少药物剂量可以降低毒性,而毒性对这些药物能否获批非常关键。
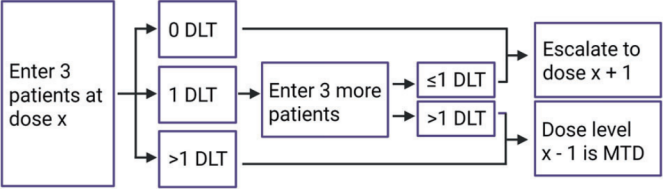
表1.“3 + 3”一期试验设计(DLT:剂量限制毒性;MTD:最大耐受剂量)[1]
药物开发策略的目的,是在药物靶点上实现最大的生物效应,从而转化为治疗效果。因此,对精确评估药物作用机制和药效学作用的分子生物学标志物的需求非常大。
在临床试验中,纳入这些标志物可以证明:(i)机制,即药物达到预期目标的证据,(ii)概念验证,即药物击中靶点并改变肿瘤生物学的证据,(iii)确定最佳生物剂量,以及(iv)对缓解率/耐药机制的理解。
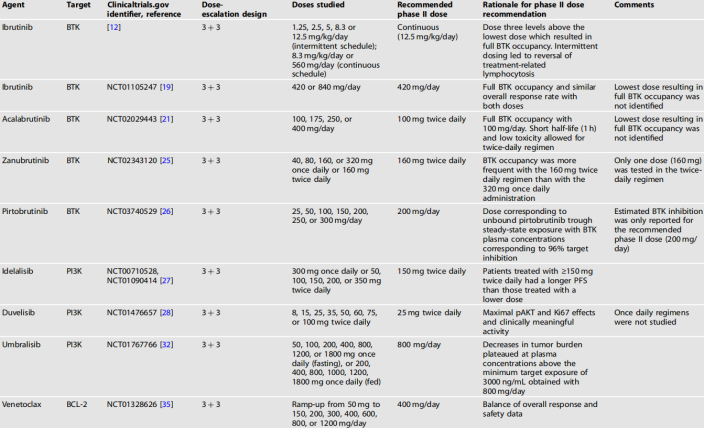
图1 CLL靶向治疗的I期试验[1]
在传统的“3 + 3”临床I期试验设计中,最初用于研究细胞毒药物,使用剂量限制性毒性(DLT)而不是药理学生物标志物来指导剂量递增。3名患者首先被纳入指定剂量队列。在没有任何DLT的情况下,另外3名患者被纳入更高剂量队列。如果队列中有1例患者发生DLT,则另外3例患者被纳入同一剂量队列。如果没有其他患者发生DLT,则该剂量被定义为最大耐受剂量(MTD)。如果6例患者中有2例或2例以上发生DLT,则已经超过MTD(表1)。
值得注意的是,MTD在治疗的第一个周期就已经确定。分子靶向治疗需要比细胞毒药物更长的治疗方案,治疗紧急毒性可能出现在治疗过程的后期。虽然已经提出了替代“3 + 3”临床I期试验设计的方案,但“3 + 3”设计仍常用于慢性淋巴细胞白血病(CLL)试验(图1)。
2008年,Methodology for the Development of Innovative Cancer Therapies (MDICT)提出,MTD和药代动力学是确定靶向药物剂量的合理I期终点。这一建议的理由没有明确指出,但似乎是基于对31种靶向药物的57项I期试验的回顾,这些试验表明毒性是停止剂量递增和确定进一步研究的剂量的最常见决定因素。这些建议在CLL靶向治疗的剂量探索研究中已经不同程度地遵循。
下文回顾了目前已批准CLL疗法的I期研究中,各自的剂量选择策略。从每项研究中,收集了有关起始剂量、剂量递增方法和确定推荐的II期剂量的数据,并讨论在靶向治疗方面的初步临床经验:如何将剂量减少、间歇给药和组合用药作为克服治疗不耐受和耐药的策略。
一、CLL靶向治疗的剂量探索研究
1、BTK inhibitors
在一项针对复发/难治性(R/R) B细胞非霍奇金淋巴瘤(NHL)和CLL的ibrutinib(BTKi)的I期开放标签、剂量递增研究中,确定推荐的II期剂量时,使用了共价抑制剂ibrutinib对BTK激酶的占有水平作为生物标志物。这是基于一项对犬的研究结果,该研究证明了治疗疗效与BTK占有水平之间的相关性。
在I期试验中,患者口服伊布替尼,每天1次(无一例死亡;OD),1.25、2.5、5、8.3或12.5 mg/kg,连续28天,7天休息;或连续服用8.3 mg/kg或560 mg/d。在第一个周期结束,评估DLT后开始剂量递增(35天)。
MTD被定义为≥33%的患者经历DLT的剂量,或高于最低剂量3个剂量水平,前提是BTK激酶被完全占用,并且未观察到DLT。将剂量增加三个水平的理由是值得怀疑的,这种设置等于默认了,将选择最高或第二高剂量,因为只测试了五种剂量。
在这项研究中,ibrutinib的MTD没有达到,整个队列的56例患者中只有2例报告了DLT。在2.5 ~ 12.5 mg/kg/d的所有剂量水平下,BTK激酶的占用>95%,缓解率相似。虽然没有明确说明,可能因激酶占用未完全饱和,II期推荐剂量仍选择了12.5 mg/kg/d。由于CLL患者在间歇给药方案的7天休息期间出现了治疗相关的淋巴细胞增多的短暂逆转,因此建议采用连续给药方案。
淋巴细胞增多是伊布替尼和其他B细胞受体抑制剂的常见副作用,表示淋巴细胞从淋巴结腔室流出。淋巴细胞增多是短暂的,与不良事件、无进展生存期较低(PFS),或疾病进展不相关。
图2 批准上市的BTK抑制剂的结构[3]
在随后的Ib-II期研究中,ibrutinib治疗85例R/R CLL患者,51例患者服用420 mg OD,34例患者服用840 mg OD。对应于70kg患者,分别为6mg /kg和12mg /kg的剂量。两种剂量下均观察到BTK的充分占用,两组的总体缓解率相同。基于此,420mg的剂量建议用于复发的CLL。这个剂量低于初始一期试验的推荐剂量。
2014年,FDA批准ibrutinib加速批准用于之前至少接受过一次治疗的CLL患者。2016年,ibrutinib被批准为CLL的一线治疗,在RESONATE-2研究中,将其与苯丁酸氮芥进行比较。2019年,FDA批准使用伊布替尼与obinutuzumab(抗cd20抗体)联合治疗之前未治疗的CLL成年患者,并在2020年扩大适应症,包括其与利妥昔单抗的联合(抗cd20抗体)用于CLL的一线治疗。它也得到了EMA的批准。推荐的伊布替尼剂量为420 mg口服OD。
Acalabrutinib是第二代共价BTKi。与依布替尼相比,它的选择性更强,脱靶效应更少,不良事件也更少。在61例复发CLL患者的I-II期研究中,在研究的剂量递增部分acalabrutinib的剂量为100-400 mg。在最低剂量下已观察到BTK激酶完全被占用(99-100%),因而选择了100mg的剂量。acalabrutinib的半衰期为1 h,而ibrutinib的半衰期为4-13 h。基于这些考虑,再加上acalabrutinib的低毒性,允许在II期研究中每天给药两次。

图3 部分批准的靶向治疗慢性淋巴细胞白血病[1]
基于ELEVATE-TN和ASCEND的研究,2019年FDA批准acalabrutinib用于CLL的治疗。该药物于2020年被EMA批准为之前经过治疗的CLL的单一疗法。推荐剂量为每12小时口服100mg。
Zanubrutinib是另一种选择性共价BTKi。在一项关于zanubrutinib治疗R/R B细胞恶性肿瘤的I期研究中,患者接受40、80、160或320 mg OD,或160 mg 两天一次;所有剂量的中位数BTK激酶占用率均>95%,但160 mg BID组比320 mg OD组的BTK持续占用率更高,这也是II期剂量选择160mg BID的原因。扩大研究范围,加入40或80mg BID方案是很有意义的,因为这些剂量在给予OD时,表现与160mg OD方案一样好。
Zanubrutinib是由FDA批准的套细胞淋巴瘤的治疗(2019年)和Waldenström的巨球蛋白血症(2021年),但还没有用于CLL。160mg BID和360mg OD剂量都已被FDA批准。
Pirtobrutinib是在研的第一代非共价BTKi,其对BTK C481突变的CLL也有效。在一项I期研究中,pirtobrutinib用于323例B细胞恶性肿瘤患者,给药剂量为25,50,100,150,200,250或300mg OD,未观察到DLT。根据96%的预估靶标抑制率[4],II期推荐剂量设置为200 mg/d。其他剂量的靶标抑制率未见报道。
2、PI3K inhibitors
Idelalisib是一种PI3K抑制剂(PI3Ki),更特异地阻断p110δ。在一项I期研究中,54例R/R型CLL患者接受idelalisib 300 mg OD或50、100、150、200、350 mg BID治疗。由于OD给药不能与BID给药保持相同的连续血浆暴露水平;此外,BID≥150 mg的患者比低剂量治疗的患者有更长的PFS(分别为32个月和7个月)。
基于此,推荐150 mg BID作为II期剂量。Idelalisib于2014年获得EMA和FDA批准用于CLL,推荐剂量为150mg BID。
Duvelisib是下一代的双p110γ/δ PI3Ki。在一项I期剂量递增研究中,31例晚期血液恶性肿瘤患者接受了8、15、25、35、50、60、75、100 mg duvelisib BID治疗。duvelisib的半衰期为5.2 ~ 10.9 h,与ibrutinib相似。ibrutinib的长半衰期是不测试该药物的BID方案的原因,那么,为什么不研究duvelisib的OD方案呢?因为根据DLT的发生情况,MTD被确定为75mg BID。
PI3K信号传导抑制(pAKT)和增殖抑制(Ki67)不受剂量依赖,在25 mg BID时最高。这项研究扩大到179名患者,他们接受了duvelisib BID为25或75 mg。≥3级AEs和总缓解率在两个队列中相似。基于这些发现,推荐更低的25mg BID作为II期剂量。
图4 上市的PI3K抑制剂的结构[5]
2018年,FDA批准Duvelisib用于至少之前接受过两次治疗的CLL患者。该批准基于DUO研究,其中duvelisib与抗cd20抗体ofatumumab进行了比较。随后在2021年获得了EMA的批准。推荐剂量为25mg BID。
Idelalisib和duvelisib都有严重的毒性,这导致对这两种药物添加黑框警告。此外,由于无法完成验证性试验,这些药物的开发商最近主动撤销了滤泡性淋巴瘤(FL)的加速批准。
Umbralisib是p110δ选择性PI3Ki。在一项针对R/R CLL和淋巴瘤患者的umbralisib的I期研究中,患者接受了50、100、200、400、800、1200、1800mg OD的剂量。另外的队列接受200、400、800、1000、1200、1800mg OD(微粉剂型)。umbralisib的半衰期超过100小时。在接受了1800 mg/d的微粉配方的患者中报告了两例DLT。MTD因此被确定为1200 mg/d。当剂量为800或1200 mg时,umbralisib的血浆浓度均高于最低目标暴露量3000 ng/mL(5.25µM)。当血浆浓度超过3000 ng/mL时,肿瘤负荷下降趋于平稳。基于这一发现,推荐800 mg/d为II期剂量。
Umbralisib于2020年获得FDA快速审批状态,与抗cd20抗体ublituximab联用治疗CLL,并于2021年获得FDA批准用于FL和边缘区淋巴瘤(MZL),推荐剂量为800mg OD。
然而,对6项PI3Ki治疗惰性NHL或CLL的随机对照试验的分析导致了对PI3Ki组总生存率较低的担忧,以及随后umbralisib因已批准的适应症却自愿退出市场,并撤销umbralisib联合ubluximab治疗CLL和SLL的申请。FDA的一项评论也强调了PI3Ki的剂量选择需要谨慎。
3、Venetoclax(BCL-2拮抗剂)
Venetoclax是一种BCL-2拮抗剂。在一项venetoclax的I期研究中,患者首先接受了20 mg或50 mg的试验剂量,以检测肿瘤溶解综合征的发生。患者随后接受了venetoclax,经过3周的递增计划,最终剂量为150、200、300、400、600、800或1200 mg OD。扩展队列在5周后从20mg OD开始,接受了400mg的最终剂量。50 mg剂量后,venetoclax的半衰期约为19 h,在所有研究剂量下均有活性。对于接受<400、400和>400 mg治疗的患者,15个月时的PFS分别为58%、69%和77%。II期推荐剂量400mg OD是根据缓解率和安全性数据确定的,但没有提供进一步的细节。
2016年,FDA批准venetoclax用于经过治疗的del(17p) CLL患者。根据MURANO研究,一项比较venetoclax +利妥昔单抗与苯达莫司汀+利妥昔单抗治疗R/R CLL患者的随机III期临床试验,FDA于2018年批准了venetoclax用于之前至少接受过一次治疗的CLL患者,无论有无del(17p)突变。CLL14研究比较了venetoclax + obinutuzumab和chlorambucil + obinutuzumab在之前未治疗的CLL患者中的疗效。中位随访28.1个月后,预计24个月的PFS在venetoclax组为88.2%,obinutuzumab组为64.1%。
基于这项研究,FDA于2019年批准了venetoclax用于所有CLL患者。推荐的剂量是5周内从20mg OD增加到400mg OD。
二、降低剂量的靶向治疗未必会影响疗效
“3 + 3”一期试验设计(图1)的一个伦理问题是,一些患者可能使用次优剂量进行治疗。然而,一项对24项I期试验中683例使用低于、在或高于MTD剂量治疗的患者的系统分析表明,使用低剂量靶向治疗的患者并不一定比试验中的其他患者表现出更差的结果。
这一发现也表明,靶向治疗与细胞毒药物有不同的剂量-效应关系。靶向治疗药物的毒性较轻,往往在达到MTD前已经达到靶点饱和,发挥了最大的肿瘤抑制作用。
一项临床试点研究调查了ibrutinib在三个28天周期内的药代动力学和药效学影响,从推荐剂量420 mg/天,通过280 mg/天减少到140 mg/天。研究表明,BTK占用、抑制BTK下游信号传导和自磷酸化(Tyr223)、以及降低血浆趋化因子CCL3和CCL4的水平,在三个剂量水平上相似,表明如果伊布替尼的效果达到目标,则目前建议的剂量过高。
为了支持这些发现,一些回顾性研究表明,临床上表明减少伊布替尼剂量并不影响CLL的预后。此外,在一项TP53异常的CLL前瞻性研究中,伊布替尼剂量强度不影响PFS。然而,虽然伊布替尼剂量强度不影响患者的预后,忘记服用伊布替尼可能会。
一项对III期临床试验的回顾性研究显示,伊布替尼连续缺失≥8天的患者的中位PFS比缺失<8天的患者更短,一项对英国和爱尔兰315例患者的回顾性研究显示,与整个队列相比,第一年治疗内,停用伊布大于14天的患者减少了1年的总生存期(68.5% vs. 83.8%)。然而,剂量中断和剂量改变不影响经venetoclax治疗的CLL患者的PFS。
三、从PI3K抑制剂毒性学到的经验—间歇给药
PI3Ki对CLL有效,但与第一代抑制剂idelalisib和duvelisib相关的严重毒性限制了它们的使用。据报道,长期暴露于这类靶向治疗会增加不良事件发生率,这使得传统3 + 3设计的I期研究具有挑战性,因为剂量是在第一个治疗周期后确定的。因此,应考虑这些抑制剂的替代试验设计
尽管如此,我们从PI3Ki的临床经验中获得的经验已经允许开发更特定的下一代抑制剂和优化治疗方案。克服治疗毒性的一种策略是从连续给药改为间歇给药。
一项关于idelalisib +利妥昔单抗治疗CLL的回顾性研究表明,治疗获益远远超过治疗时间(中位PFS 29.6个月,中位治疗时间11.9个月)。这一发现证实了在前瞻性临床试验中对idelalisib的时间限制或间歇给药的研究。这两种PI3Ki copanlisib和zandelisib的替代给药方案已经建立,并正在研究其他药物。
Zandelisib是新一代p110δ抑制剂,其p110δ占用时间比idelalisib更长。在一项对健康志愿者进行的zandelisib的I期研究中,基于对嗜碱性粒细胞活化的高度抑制,确定60 mg OD为推荐的II期剂量。在FL和CLL/SLL中进行的一项Ib期zandelisib研究显示,最常见的AEs发病延迟超过第2周期。这些AEs可以通过中断治疗逆转。
这些发现推动了在两个连续周期后进行间歇给药(7天/21天)的I期试验。这段时间的基本原理是基于调节性T细胞重新繁殖所需的时间。初步结果表明,间歇给药保持了疗效,但降低了延迟3级AEs的发生率。II期TIDAL试验旨在比较R/R滤泡性淋巴瘤患者的连续和间歇给药方案,但已修订为仅研究间歇给药(NCT03768505)。正在进行的COASTAL III期研究仅研究zandelisib (NCT04745832)在复发性惰性NHL患者中的间歇治疗方案。在2020年,FDA批准Zandelisib快速通道用于治疗至少接受过2种系统治疗的成人R/R FL患者。
Parsaclisib是另一种下一代p110δ抑制剂。在R/R B细胞恶性肿瘤患者的I期试验中,研究了间歇给药。前9周服用Parsaclisib 20mg OD,随后每周服用20mg以减少迟发AEs。本设计基于p110α/β/γ/δ抑制剂copanlisib的药代动力学和药效学比较模拟。没有报告因间歇给药臂发生AE而中断治疗,而连续给药臂有13%的患者中断治疗。High-grade的AEs在间歇给药组中也较少。Ib/IIa期topMIND试验正在研究在R/R CLL (NCT04809467)中间歇给药parsaclisib联合tafasitamab(抗cd19抗体)。
这些研究表明,间歇给药是克服PI3Ki耐受不良的一种策略。这种策略现在也在研究duvelisib。在II期TEMPO CLL/SLL试验(NCT03961672)中,duvelisib首先连续给药3个周期,然后在每个周期的第1-2、8-9、15-16、22-23天给药。
四、借助生物标志物确定临床剂量选择
对CLL靶向治疗的剂量探索研究的回顾表明,传统的“3 + 3”设计仍然大量用于CLL的新药物。由于DLT的测定与靶向药物的相关性小于细胞毒药物,因此在每次试验中都需考虑额外的因素。
然而,只有BTKi对药物靶点(BTK占用)的直接影响在多个试验中得到了一致的研究。对于PI3Ki,已经证明很难确定一个终点。当一个合适的分子靶向终点可用时,就像BTKi的情况一样,使用这个来指导剂量选择可能表明比毒性驱动设计获得的剂量更低。
该发现强调了这一点,即在本文列举的临床试验中,导致BTK完全占用的最低BTKi剂量要么没有确定(测试的最低剂量导致BTK完全占用),要么没有报告(只报告了推荐的II期剂量)。这表明仍有空间来优化这些药物的剂量,这与许多报告一致,这些报告表明临床上减少伊布替尼目前批准的剂量不会影响CLL的结果。
靶向治疗的联合方案作为一种提高缓解率和克服耐药性的策略在CLL中越来越重要。这意味着患者可能会经历多种药物的副作用。在推荐II期药物剂量时应考虑到这一点,因为较低的药物剂量可能导致较低的毒性。联合使用的药物可能产生协同作用,这将扩大它们各自的贡献。
这进一步证明了降低药物剂量的合理性。体外用ibrutinib + venetoclax治疗CLL细胞表明,药物协同作用发生在远低于推荐治疗剂量的剂量。CORAL研究(NCT05209308)调查了zandelisib与venetoclax和rituximab在R/R CLL中的联用,包括一项减少venetoclax剂量的初始I期研究,证明目标药物的推荐剂量是需要持续评估的。
综上所述,传统的毒性驱动的“3 + 3”一期试验设计在靶向治疗时代仍占主导地位。虽然在大多数临床试验中都研究了药效学生物标志物,但这些生物标志物在确定II期剂量时并未显示其重要性。如果更严格地使用药效学生物标志物来指导剂量的选择,那么在大多数情况下,推荐剂量将低于目前的推荐剂量。
与此同时,减少剂量也可以降低患者和卫生保健系统所经历的与治疗相关的毒性和经济压力。因此,靶向治疗药物的临床剂量应更多的参考最佳生物学剂量。当然,一个合适的药效学生物标志物也并不好确定。
参考文献:
1, Sigrid S. Skånland et al; Determining drug dose in the era of targeted therapies: playing it (un)safe? Blood Cancer Journal (2022) 12:123.
2,节选自《肿瘤专科药师临床工作手册》.
3,Tiantai Zhang et al; Emerging small-molecule inhibitors of the Bruton’s tyrosine kinase (BTK): Current development, European Journal of Medicinal Chemistry 217 (2021) 113329.
4, Mato AR, Shah NN, Jurczak W, Cheah CY, Pagel JM, Woyach JA, et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): a phase 1/2 study. Lancet. 2021;397:892–901.
5,Minhang Xin et al; Research advances on selective phosphatidylinositol 3 kinase δ (PI3Kδ) inhibitors. Bioorganic & Medicinal Chemistry Letters 30 (2020) 127457.

“精准药物”公众号由暨南大学药学院张章博士创建。张章博士主要从事:1)小分子靶向抗肿瘤药物的发现、作用机制和成药性研究;2)基于化学蛋白质组学的疾病和药物靶标发现研究。作为主要完成人,团队开发的4个激酶抑制剂先后转让给企业进行后期开发,其中1个已上市(奥雷巴替尼),1个报生产,1个在I期临床,1个在临床前研究阶段。实验室具备完善的体内外药效、机制、成药性、靶标发现和确证等技术平台;支撑和推动了多个企业靶向抗肿瘤药物的发现、成药性评价、新适应症和机制等研究。

