Circ Res 浙江大学余路阳团队阐明SUMO化修饰在缺氧性肺动脉高压中的关键调控机制
时间:2023-08-24 13:42:12 热度:37.1℃ 作者:网络
肺动脉高压是一类难治性、高致死性的疾病,深入研究其发病机制与病理过程,对研发靶向药物与诊疗方式具有重要的指导意义。缺氧性肺动脉高压是肺动脉高压(PH)的主要类型,由于肺动脉压力和肺血管阻力的增加,最终导致右心室衰竭和死亡,是一类典型的肺血管重塑性疾病。临床上,慢性缺氧是缺氧性肺动脉高压的主要诱导剂和促进剂,而肺动脉氧分压的降低进一步促进肺动脉高压的发展。
尽管已有大量研究表明,缺氧诱导因子(HIFs)信号在早期缺氧中对血管系统起着重要调控作用,然而肺动脉系统如何响应长期持续性缺氧的机制尚不明确。
血管是由内皮细胞、平滑肌细胞、成纤维细胞和脂肪细胞等共同组成的复杂、且具有自分泌和旁分泌功能的组织器官。内皮细胞作为血液与血管壁的交界面,是感知和响应血流动力学、体液以及血管缺氧刺激的前线,并将反应性信号传递给血管中膜及外膜。因此,内皮功能障碍是大多数血管重塑和疾病的常见初始事件。内皮缺氧介导的内皮功能障碍被认为是缺氧性肺动脉高压和其他缺氧性血管疾病的主要原因。
线粒体是细胞感应并消耗氧气的重要场所,大量研究表明线粒体功能障碍会导致心血管细胞的细胞过程停滞,尤其在心肌细胞、血管平滑肌细胞和血管周围脂肪细胞等含有较高水平线粒体的细胞中(占细胞质体积30%-35%)。然而,血管内皮细胞中线粒体仅占细胞质体积的2%至6%,且内皮细胞中的ATP主要来源于无氧糖酵解,而非线粒体氧化磷酸化。因此,内皮线粒体被认为是信号细胞器而非能量供应器。内皮线粒体作为信号细胞器在心血管发育和功能维持中的重要作用被逐渐重视。线粒体是高度动态的细胞器,通过调整分布、动力学平衡和细胞器间通讯来响应微环境的变化。
SUMO化(类小泛素化)修饰是一类重要的蛋白翻译后修饰,SUMO分子分别在E1/E2/E3级联酶和SENP特异性去SUMO化蛋白酶(SENPs)的调控下与靶标蛋白动态偶联和分离。在哺乳动物细胞中,SENP介导的去SUMO化在维持生理功能方面发挥着重要作用,SENP1介导的去SUMO化在缺血和血管缺陷中起重要作用,且SUMO化修饰在移植性动脉硬化中的内皮激活和血管生成中内皮生长具有关键作用。临床和动物研究表明,缺氧缺血应激增加了脑和心脏血管系统中SUMO化共轭水平,并在线粒体中有显著的定位,提示线粒体中的SUMO化修饰在相关血管病变中的潜在作用。越来越多的研究者注意到线粒体重塑的病理生理功能与内皮线粒体介导的功能密切相关,在心血管发育和功能维持中起着重要作用,但是关于内皮线粒体在血管稳态中的功能机制仍不清楚。
2023年8月17日,浙江大学生命科学学院余路阳团队在心血管领域顶尖杂志Circulation Research上发表题为“Endothelial FIS1 DeSUMOylation Protects Against Hypoxic Pulmonary Hypertension”的研究论文,首次阐明了线粒体蛋白FIS1的去SUMO化修饰通过维持内皮线粒体稳态,适应性地保护肺动脉功能和抑制缺氧性肺动脉高压;肺动脉内皮中FIS1去SUMO化-SUMO化转变是缺氧性肺动脉高压的重要发病机制。该研究为缺氧性肺动脉高压等相关缺氧性心血管疾病的预防和治疗提供了新思路。

首先,作者通过研究处于不同临床病理阶段的缺氧相关肺动脉高压(PH)患者的肺组织标本及非PH肺组织标本为出发点,确定SENP1介导的SUMO化修饰与肺内皮线粒体重塑相关联,并通过序列预测及实验筛选出关键线粒体蛋白FIS1的SUMO化修饰位点。进一步通过检测来自非PH供体和不同病理阶段的PH患者的肺动脉内皮细胞和不同低氧时间下的人肺动脉内皮细胞中的SENP1表达分布与FIS1的SUMO化修饰水平,明确了短期缺氧下,肺内皮SENP1由细胞核易位至内皮线粒体中从而增强线粒体蛋白FIS1的去SUMO化,进而阻止线粒体过度分裂;而长期缺氧下,由于SENP1的缺失而导致FIS1的过度SUMO化修饰是破坏线粒体动力学平衡的关键因素。
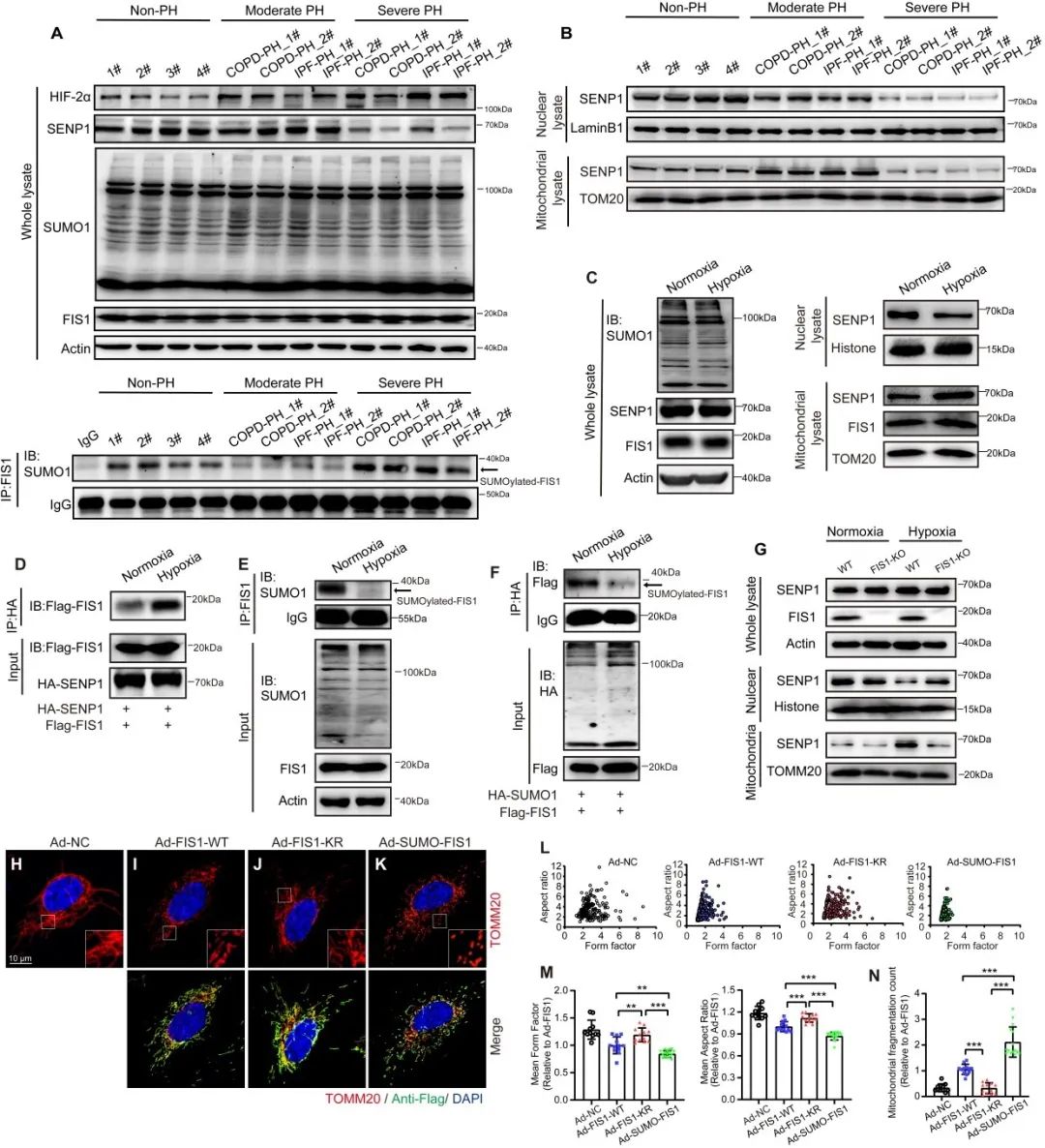
图1. 短期低氧条件下,SENP1(特异性蛋白酶1)转移至内皮线粒体中,增强了FIS1(线粒体分裂蛋白1)的去SUMO化修饰
为进一步探索内皮线粒体FIS1的去SUMO化-SUMO化修饰在内皮细胞中的调控作用,研究团队基于质谱的蛋白质组学分析发现,去SUMO化修饰的FIS1 (FIS1-KR)与更多的线粒体-内质网膜偶联结构(MAM)相关蛋白有相互作用。进一步通过线粒体-内质网膜偶联结构(MAM)的亚细胞器分离技术、免疫共沉淀、透射电镜及成像、钙成像/电流、流式细胞术、细胞耗氧率(OCR)和细胞外酸化率(ECAR)监测等研究手段,发现SENP1介导的FIS1去SUMO化修饰(FIS1-KR)促进FIS1和MFN2(线粒体融合蛋白2)和VDAC1(电压依赖性阴离子通道1)蛋白间的相互作用,从而保持线粒体的动力学平衡、MAM的形成和细胞内钙稳态。
机制上,短期缺氧诱导SENP1易位到线粒体,而SENP1通过靶向FIS1来调节内皮线粒体的重塑过程。在长期持续性缺氧时,肺血管内皮线粒体SENP1的缺失直接导致FIS1过度SUMO化修饰,线粒体动力学平衡向过度分裂转变,并破坏了MAM的形成,进而引发线粒体功能障碍和内皮细胞代谢重编程,并最终导致肺内皮功能障碍和肺动脉高压的发展。
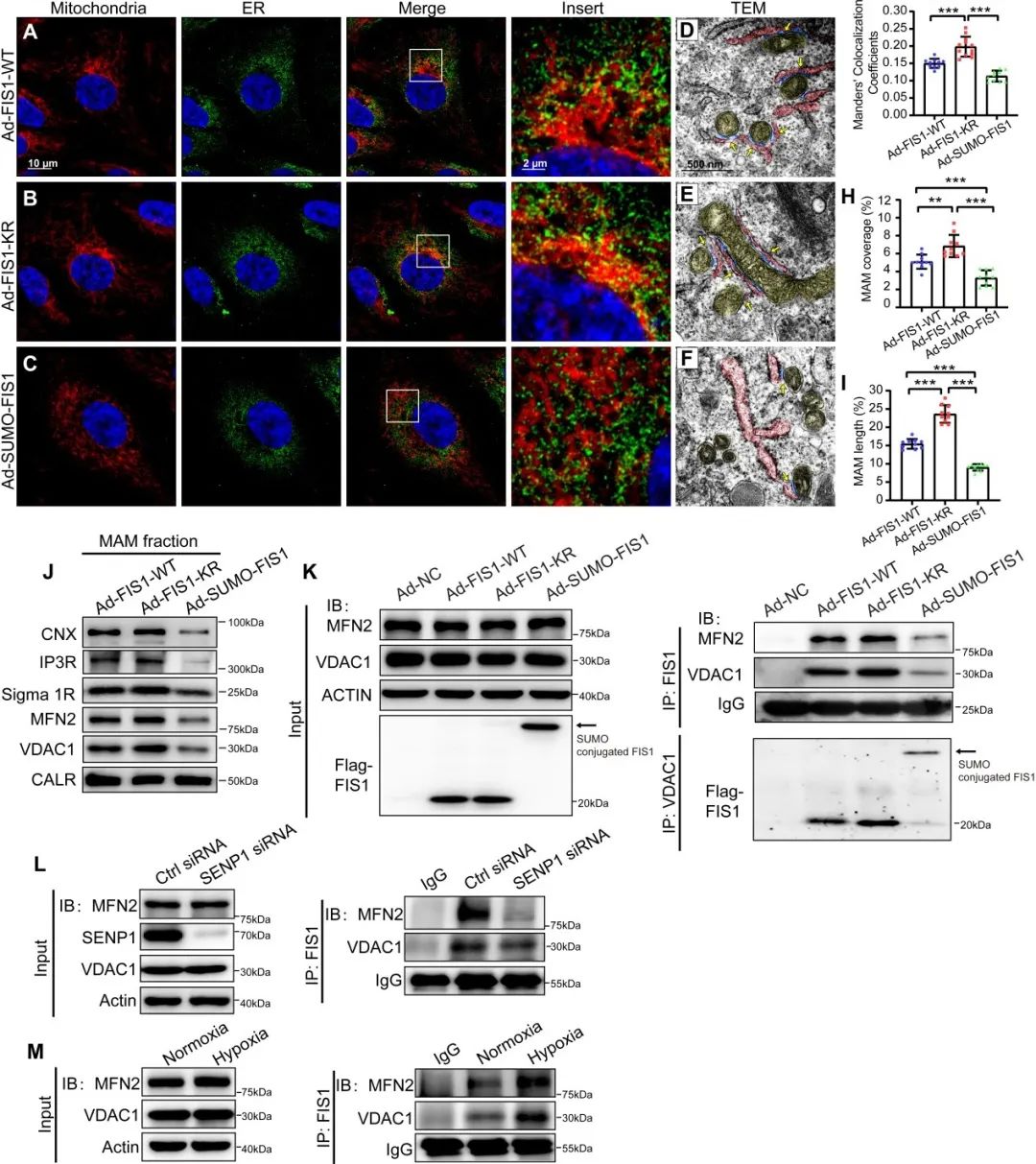
图2. FIS1的去SUMO化修饰通过增强FIS1与MFN2(线粒体融合蛋白2)的相互作用而调控细胞器间的通讯
最后,研究团队根据PH临床样本、大鼠肺动脉高压模型、内皮特异性SUMO-FIS1/FIS1-WT敲入小鼠的缺氧性肺动脉高压模型及低氧培养的人肺动脉细胞中的实验观察,证明在短期缺氧下,FIS1的过度SUMO化修饰促进线粒体分裂与内皮损伤,最终加剧肺血管重构与PH发展。而FIS1的去SUMO化修饰通过维持线粒体动力学平衡与MAM稳态,进而保护了线粒体代谢与内皮功能;且在缺氧性PH小鼠中,通过气道灌注携带FIS1的SUMO化突变体(FIS1-KR)腺相关病毒(AAV-FIS1-KR),能有效保护线粒体动力学平衡与血管功能,缓解血管重构的进程,并最终延迟缺氧性PH的发展。
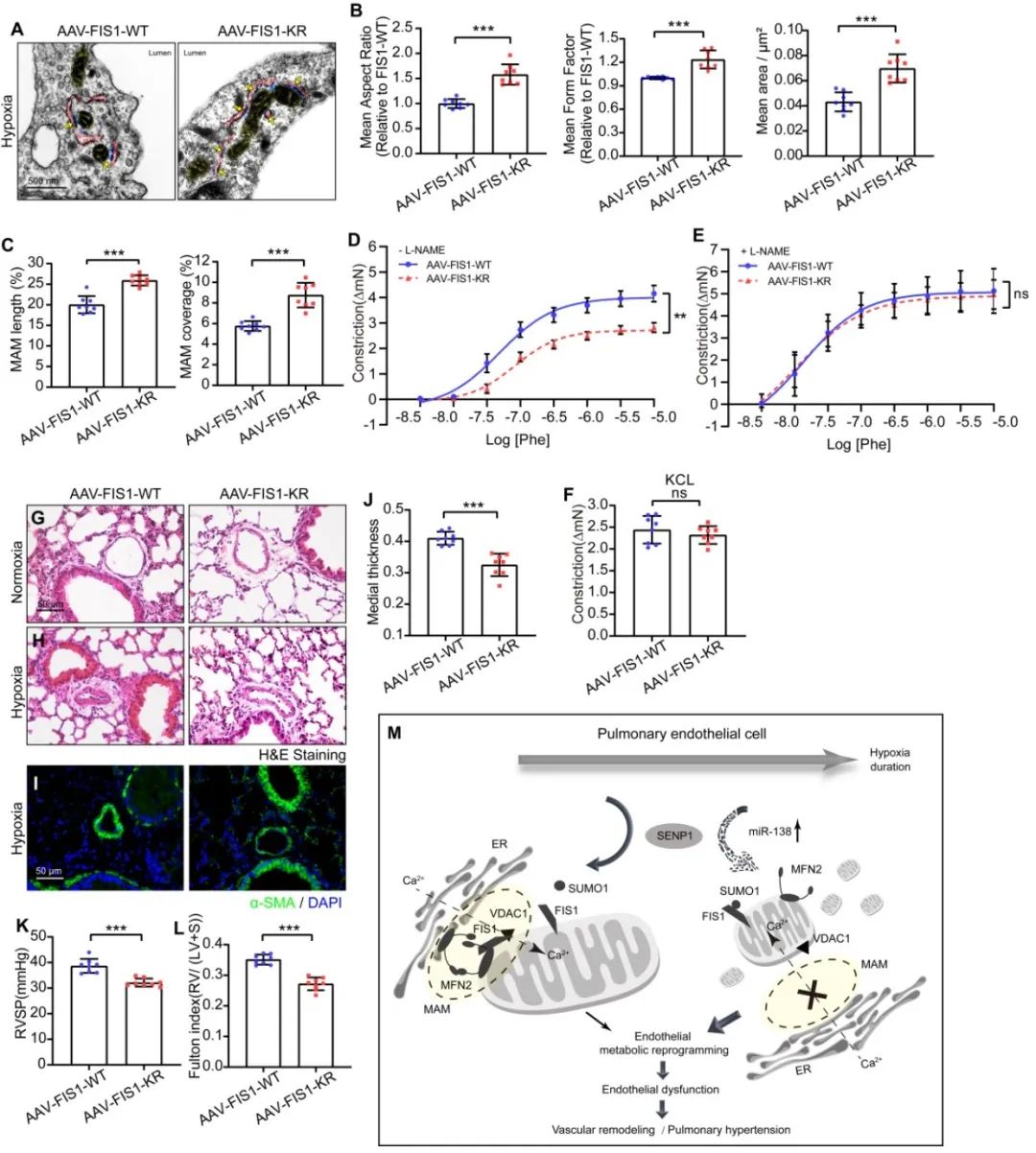
图3. 内皮特异性引入去SUMO化的FIS1(FIS1-KR)有助于缓解缺氧诱导的肺动脉高压的发展
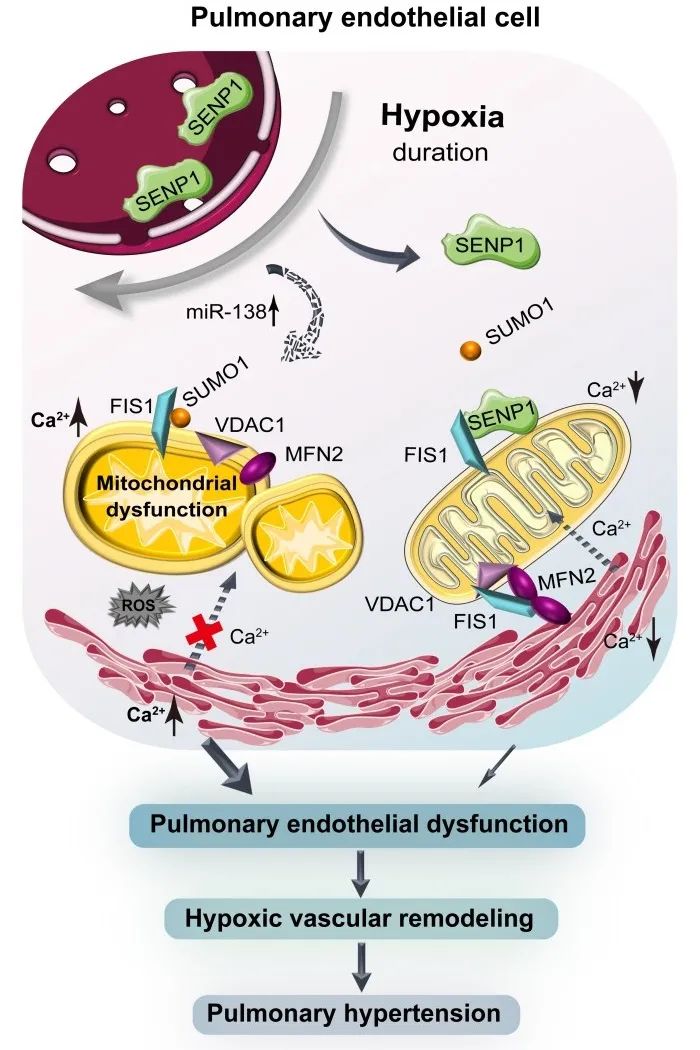
综上,本研究揭示了短期缺氧下,内皮SENP1的线粒体易位介导的FIS1的去SUMO化修饰维持了线粒体动力学平衡与结构完整性,以及MAM中的内质网-线粒体间的钙通讯,从而保持肺内皮细胞功能和血管稳态。与之相反,长期缺氧通过诱导miR-138而抑制SENP1的表达及线粒体中SENP1的可用性,线粒体FIS1的过度SUMO化修饰导致肺内皮线粒体功能障碍和内皮代谢重编程,进而加剧内皮功能损伤与PH发展。因此,肺内皮SENP1介导FIS1的去SUMO化-SUMO化修饰在不同缺氧阶段时的转换是缺氧性PH的重要病理机制。
本研究由浙江大学生命科学学院余路阳教授实验室为主完成,工作受到了浙江大学医学院附属邵逸夫医院心内科、第一医院心内科、转化研究院,浙江省中医药大学基础医学院,北京大学基础医学院、耶鲁大学医学院等研究团队的协助和支持。浙江大学生命科学学院博士生周晓菲和蒋苑青为本文共同第一作者,余路阳教授为本文通讯作者。本研究受到国家重点研发专项、国家自然科学基金项目以及浙江大学创新团队项目和世界顶尖大学合作计划项目等的资助。
原文链接:
https://www.ahajournals.org/doi/10.1161/CIRCRESAHA.122.321200?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed

