NTRK1融合胰腺腺泡细胞癌化疗进展,拉罗替尼无效!与NBN回复突变有关?
时间:2024-06-19 16:00:21 热度:37.1℃ 作者:网络
胰腺腺泡细胞癌(PACC)是一种罕见的胰腺癌,通常具有可靶向的变异,包括MAPK通路中的激活性融合和DNA损伤应答/同源重组DNA修复相关基因的功能缺失(LOF)变异。本文描述了一名同时携带NBN体细胞双等位基因LOF和激活的NTRK1融合的PACC患者。在接受亚叶酸、氟尿嘧啶、伊立替康和奥沙利铂(FOLFIRINOX)治疗13个月后出现疾病进展时,转移性肝活检的基因组分析显示出现了恢复NBN阅读框架的新型回复突变。据研究者所知,NBN的基因回复在任何类型的肿瘤中还没有被报道为耐药机制。患者接受了选择性NTRK抑制剂靶向治疗,但无反应。该病例突出了PACC复杂但具有高度可干预性的基因谱,并强调了对罕见肿瘤类型(如PACC)进行基因组分析的价值。
研究背景
胰腺腺泡细胞癌(PACCs)是胰腺癌的罕见类型,约占成人胰腺肿瘤的1-2%,占儿童胰腺肿瘤的15%。PACCs在男性中更为常见,男女比例为2∶1。组织学上,肿瘤细胞具有腺泡细胞分化的特征,胞质中含有中等数量的嗜酸性粒细胞和PAS阳性的抗淀粉酶原颗粒,细胞核均匀,有明显的核仁,呈腺泡、腺样、实性或小梁样排列。在基因组学上,PACCs有不同于胰腺导管癌的一系列变异,很少表现出KRAS、TP53、CDKN2A和SMAD4体细胞变异,更常见的是携带影响BRAF、RET、RAF1和NTRK1/2/3的频繁激活性融合,多达30%的患者存在这些融合。此外,一部分PACCs患者携带影响DNA损伤反应(DDR)/同源重组(HR)DNA修复相关基因的胚系致病性变异,包括ATM、BRCA1、BRCA2和PALB2。
PACCs患者的主要治疗方法是切缘阴性的手术切除,然后进行化疗。目前,美国食品和药物管理局还没有批准专门用于PACC患者的靶向治疗。肿瘤携带致癌融合的患者可使用带有肿瘤类型不可知标签的疗法进行治疗,例如用于NTRK融合的恩曲替尼或拉罗替尼,或者用于RET融合的塞普替尼。聚(ADP-核糖)聚合酶(PARP)抑制剂已被批准用于携带胚系BRCA1或BRCA2致病突变的胰腺癌(包括PACC)患者,目前正在开展进一步研究,探讨PARP抑制剂在因其他经典HR相关基因变异而导致HR缺陷(HRD)的胰腺癌(NCT04858334)和未选择的PACC(NCT05286827)中的益处。
nibrin基因(NBN,也称为NBS1 [Nijmegen断裂综合征1])是DDR/HR DNA修复机制的重要组成部分。胚系NBN功能缺失(LOF)变异不仅与Nijmegen断裂综合征有因果关系,而且已被证明可导致多种癌症类型的风险增加。体细胞致病性NBN变异对胰腺癌发生和进展的影响尚不确定。
本报告描述了1例PACC患者,其肿瘤同时具有激活性SEL1L::NTRK1融合和NBN体细胞双等位基因失活。在接受亚叶酸、氟尿嘧啶、伊立替康和奥沙利铂(FOLFIRINOX)治疗后出现疾病进展时,检测到一个恢复NBN阅读框的新型回复突变,这表明NBN LOF、PACC进展和对主要治疗方案的治疗耐药发展之间存在潜在联系。这种影响NBN基因的新型回复突变在越来越多的靶向DNA损伤和DDR治疗的耐药机制列表中又增加了一项。
研究结果
临床病程:
一名65岁白人男性患者因腹痛就诊。计算机断层扫描显示胆总管轻度扩张,超声内镜进一步评估显示胰头部有肿块。对该肿块进行了细针抽吸,细胞学检查结果符合腺癌(图1a)。分期评估提示pT1c、pTN2、pM0(AJCC第8版),行胰十二指肠切除术。病理检查示腺泡细胞癌,大小约2 cm,结构多样,包括实性小梁和腺泡结构,侵犯十二指肠和肝内胆管,16个区域淋巴结中有4个受累。在细胞水平,细胞质呈颗粒状,细胞核呈单形,核仁明显。免疫组化检测突触素、嗜铬粒蛋白、MOC-31/EpCAM均阴性,PAS-D检测胞浆颗粒阳性。鉴于该肿瘤类型罕见,诊断得到了多名病理医师的确认和同意。此外,研究者回顾性地发现,用于确定分子合格条件的筛查组织显示BCL-10阳性,与诊断一致。
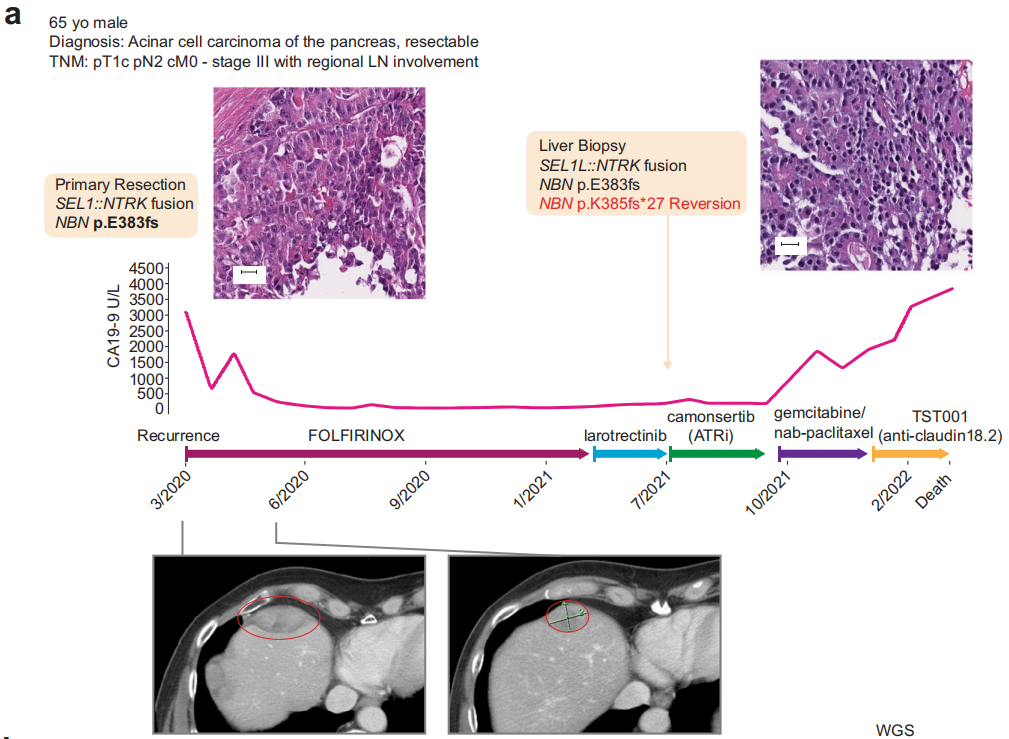
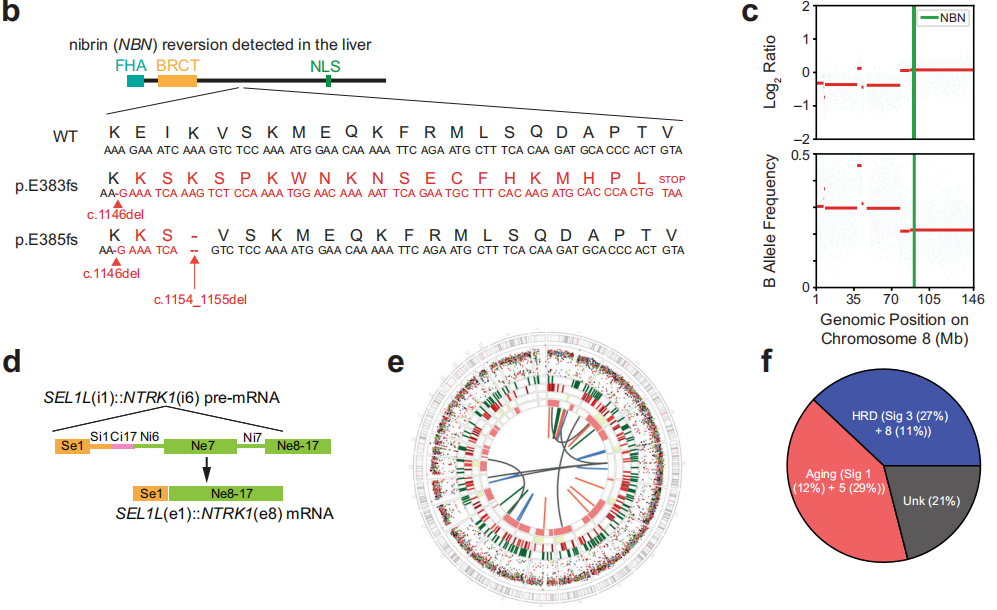

图1
随后,患者接受了吉西他滨和卡培他滨6个月的辅助治疗,随后接受了卡培他滨同步放疗。完成该治疗后,患者接受了16个月的监测,当时他被诊断为肝脏转移性疾病,组织病理学证实为转移性腺泡细胞癌(图1a)。
当时,根据标准治疗将原发性胰十二指肠切除术(本文中称为原发性切除术)的组织标本送去接受临床级别二代测序(NGS),结果发现了体细胞致病性NBN突变和框内SEL1L::NTRK1融合(表1)。患者随后接受了13个月的改良FOLFIRINOX方案姑息化疗。患者最初达到了缓解,CA19-9显著降低,肝脏肿瘤负荷降低,但最终由于新发肝脏病变和先前肝脏病变增大而停止治疗(图1a)。鉴于原发性切除标本中存在NTRK1融合,该患者接受了NTRK抑制剂拉罗替尼靶向治疗。然而,患者病情迅速进展,肝脏出现多个新病灶,既往肝脏病灶增大,导致2个月后停止治疗。
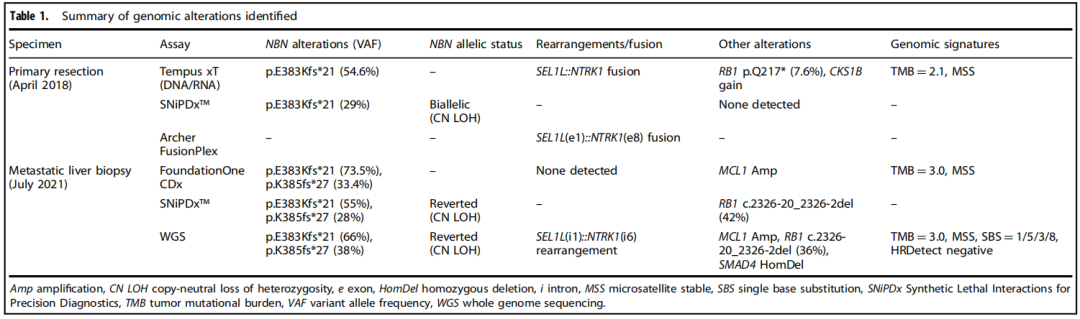
表1
此时进行了新的肝活检(在本文中称为转移性肝活检),并送去接受临床级别NGS(表1)。在等待结果期间,根据在原发性切除标本中检出的致病性NBN突变,患者被纳入TRESR(NCT04497116),并接受了治疗剂量的共济失调毛细血管扩张症和Rad3相关(ATR)抑制剂camonsertib(RP-3500,图1a)。患者在6周时疾病稳定,但12周后因靶肝脏病变进展(增大32%)而停止治疗。患者随后接受了吉西他滨和紫杉醇治疗(由于全国范围内的药物短缺,白蛋白结合型紫杉醇无法获得),2个月后由于腹膜癌恶化,疾病也出现了进展。随后,患者在一项临床试验中接受了TST001(一种抗claudin 18.2单克隆抗体)的短暂治疗,之后因临床进展而终止研究,之后患者被送入临终关怀医院,并在1个月后去世。
基因分析:
采用临床级NGS检测以及作为TRESR一部分进行的仅供研究使用的NGS检测(NCT04497116;表1)对治疗期间获得的组织样本进行了基因组分析。这包括使用SNiPDx™ panel,这是一种新型的基于NGS的诊断panel,专门用于检测和区分单等位基因和双等位基因的LOF变异,这些基因被确定为camonsertib的合成致死基因。诊断后3个月收集的原发性切除标本通过临床级NGS进行初步评估,并通过SNiPDx™进行回顾性评估,以及基于锚定多重聚合酶链反应(PCR)(AMP)的RNA测序(表1)。转移性肝活检标本在FOLFIRINOX和拉罗替尼治疗后,camonsertib开始治疗前采集,研究者通过临床级NGS进行了初步评估,并通过SNiPDx™和全基因组测序(WGS)进行了回顾性评估。
通过多种方法对组织标本和外周血单核细胞(PBMCs)进行的DNA分析发现,NBN第10号外显子存在体细胞(PBMCs不存在)致病性1-核苷酸缺失(c.1146del;p.E383Kfs*21;ClinVar变异ID:822251;表1和图1b)。这一缺失导致p.E383下游第21个密码子提前终止,预计将截断蛋白质,导致无义介导的衰变。通过SNiPDx™和WGS对等位基因特异性拷贝数的分析显示,NBN野生型等位基因的拷贝数中性杂合性丢失,随后突变等位基因重复,这与NBN的克隆体细胞双等位基因失活一致(图1c)。此外,在原发性肿瘤切除术中检测到一个亚克隆RB1(c.649 C>T;p.Q217*)致病性突变。未检测到影响TP53、ATM、BRCA1、BRCA2、PALB2或检测中包含的其他DDR/HR相关基因的胚系或体细胞致病变异。WGS、靶向DNA测序或靶向RNA测序均未检测到RET、BRAF或RAF1融合;然而,检测到一个复杂的结构重排,涉及SEL1L的外显子1和NTRK1的外显子8,导致表达的框内可能致癌的SEL1L::NTRK1融合,包含NTRK1激酶结构域(表1和图1d、e)。
转移性活检中的新型NBN回复突变:
研究者对FOLFIRINOX治疗进展后获得的肝转移活检样本和原发性肿瘤切除活检样本进行基因组比较,结果显示一个新的SMAD4纯合缺失、一个MCL1扩增和NBN中的一个第二次移码突变。该NBN突变仅在肝转移标本中检出,在原发切除标本中未检出,该突变导致初始体细胞突变下游第8个核苷酸处又出现2个核苷酸缺失(c.1154_1155del;p.K385fs*27)(表1和图1b)。对基因组测序读数的目视检查显示,NBN突变是顺式的(数据未显示),并预测将重建NBN编码框架。
NTRK1重排和融合的多模式检测:
FOLRIFINOX治疗进展后,由于存在NTRK1融合,患者接受了拉罗替尼治疗。然而,患者在开始治疗后2个月内迅速进展。鉴于靶向治疗NTRK无应答,研究者旨在验证是否存在NTRK1融合。对原发灶切除样本的靶向RNA测序证实了SEL1L::NTRK1融合的存在,并进一步确定了融合转录本的断点位于SEL1L的第1号外显子和NTRK1的第8号外显子,导致包含NTRK激酶结构域的框内融合。为了了解这一罕见分子变异的基因组机制,研究者分析了转移性肝活检的全基因组测序发现的基因组重排。SEL1L和NTRK1之间没有直接相互作用,而是两个基因与CENPC的同一内含子(内含子17)重排,导致SEL1L内含子1和NTRK1内含子6之间发生“桥接融合”(图1d、e)。这些数据表明,NTRK1外显子7的跳跃和下一个近端剪接位点外显子8的使用导致了融合转录物的高效表达。因此,靶向RNA测序和全基因组测序的联合分析阐明了这一罕见分子事件的基因组和转录后细节。在该病例中,拉罗替尼缺乏抗肿瘤活性不能用无框内NTRK1融合来解释,这提示可能涉及原发性耐药的另一种机制。
基因组特征的表征:
为了识别肿瘤的其他基因组特征,研究者仅对转移性肝活检标本进行了WGS,因为现有的原发性切除标本的肿瘤占比和剩余DNA不足。该分析显示存在8789个单核苷酸变异(SNVs)、319个插入和缺失(indels;161个插入和158个缺失)和37个结构变异(16个易位、8个缺失、8个倒位和5个串联重复;图1e)。检测发现肿瘤标本微卫星稳定,肿瘤突变负荷为3.0个突变/兆碱基,估计肿瘤倍体为1.7。主要的突变特征与衰老有关(特征1 [12%]+5 [29%]),其次是HRD(特征3 [27%]+8 [11%];图1f)。然而,该肿瘤仅显示了与HRD一致的部分基因组特征。尽管HRD相关的单核苷酸替换特征和全基因组杂合性缺失模式占主导地位,但微同源性缺失的富集程度较低,并且未观察到与HRD相关的重排特征。与这些发现一致,HRDetect评分为0.24,远低于将肿瘤分类为HRD的阈值(0.7)。这些结果与在DDR通路上游基因发生LOF变异的癌症中存在的基因组瘢痕一致。
讨 论
研究表明PACC存在DDR/HR DNA修复缺陷,但体细胞NBN缺失与胰腺癌或具体到PACC的发展没有关联。在本病例研究中,研究者对同一PACC患者的多个活检标本进行了详细的多模式基因组分析,揭示了多线治疗期间肿瘤演变的全基因组特征。
本研究结果提供了间接证据,表明体细胞NBN双等位基因突变可能在该PACC的发展和/或进展中发挥了作用。首先,NBN的双等位基因失活(由体细胞突变和拷贝数中性杂合性缺失导致)与部分HRD特征一致、但缺乏具有(微)同源性的长插入/缺失的基因组特征相关,长插入/缺失是HRDetect评分的主要驱动因素。这是意料之中的,因为HRDetect是根据乳腺癌中BRCA1/BRCA2 LOF患者的基因组进行训练的,而NBN在DNA双链断裂修复中发挥的作用位于BRCA1/BRCA2的上游位置。虽然基因组“疤痕”存在于典型的HR相关基因(如BRCA1、BRCA2、PALB2、RAD51C、RAD51D和RAD51B)中,但它们在影响DDR相关基因(如ATM和CHEK2)变异的癌症中分别不存在,但在具有较不常见HR/DDR基因(包括NBN)LOF的肿瘤中,基因组特征谱尚未完全表征。本研究结果强调了进一步研究特定HR/DDR基因LOF引起的基因组瘢痕的重要性,这不仅可能提供在没有这些基因的情况下运行的备用DNA修复机制的机制见解,而且还可能基于合成致死方法提供新的治疗机会。
其次,在初始治疗和FOLFIRINOX治疗的易感性之后,检测到另外一个预计可恢复NBN阅读框的变异。研究者根据以下情况将此解释为一种新的回复突变:(1)含铂方案FOLFIRINOX的潜在选择压力;(2)肿瘤原发NBN突变的双等位基因性质;(3)邻近性;(4)第二个NBN突变与第一个NBN突变的顺式性质;(5)在没有终止密码子的情况下,重新建立编码框架的可能性。在治疗后标本中新发现的NBN回复突变可能提示NBN功能恢复介导的适应性耐药机制。BRCA1/2、PALB2、RAD1C和RAD51D的回复变异已被证明是对含铂疗法和PARP抑制剂耐药的机制。据研究者所知,这是在任何类型的肿瘤中,铂类或PARP抑制剂治疗后NBN回复的第一份报告。
第三,尽管该患者表达了包含激酶结构域的框内NTRK1融合,但选择性NTRK抑制剂对其无应答。考虑到NTRK抑制剂在包括胰腺癌在内的各种肿瘤类型中的高缓解率,这一结果出乎意料。最值得注意的是,最近的一份病例报告描述了1例具有与本研究相同的SEL1L::NTRK1融合,但没有任何其他可能的驱动基因变异的PACC患者,该患者对NTRK抑制剂拉罗替尼有异常应答。相反,研究者可以推测,本病例中对NTRK抑制无应答的原因可能是存在NBN LOF突变和基因组不稳定,而这些与靶向治疗耐药相关。
综上所述,本研究结果与NBN LOF在PACC的进展中发挥作用一致,并提供了证据表明DDR/HR相关基因的LOF可能在PACC中发挥更广泛的致病作用。这一观察结果值得在正在进行的胰腺癌PARP抑制剂研究中考虑其他HR/DDR基因,包括PACC(NCT04858334和NCT05286827)。最后,本报告强调了对罕见肿瘤(如PACC)进行的全基因组分析所产生的独特见解和临床影响。
参考文献:
Pelster MS, Silverman IM, Schonhoft JD, et al. Post-therapy emergence of an NBN reversion mutation in a patient with pancreatic acinar cell carcinoma. NPJ Precis Oncol. 2024;8(1):82. Published 2024 Apr 1. doi:10.1038/s41698-024-00497-x

