亨廷顿病(HD):临床如何规范诊疗?
时间:2024-09-08 06:06:07 热度:37.1℃ 作者:网络
论坛导读:亨廷顿病(Huntington's Disease,HD)是由于患者大脑某些区域的神经元逐渐退化,导致不受控制的运动(即舞蹈样动作)、异常的身体姿势,以及精神行为问题和认知功能下降。亨廷顿病患者通常在30~50岁之间发病,当20岁之前出现病情时,称为青少年型亨廷顿病。亨廷顿病是由于HTT基因突变引起的常染色体显性遗传病,HTT基因编码亨廷顿蛋白,对神经系统有重要作用。当该基因1号外显子上的CAG重复次数产生异常扩增时,会导致神经元中异常蛋白的积累、正常生理功能被破坏,引发大脑纹状体及皮层等多个区域的神经细胞死亡,从而导致患者产生症状。
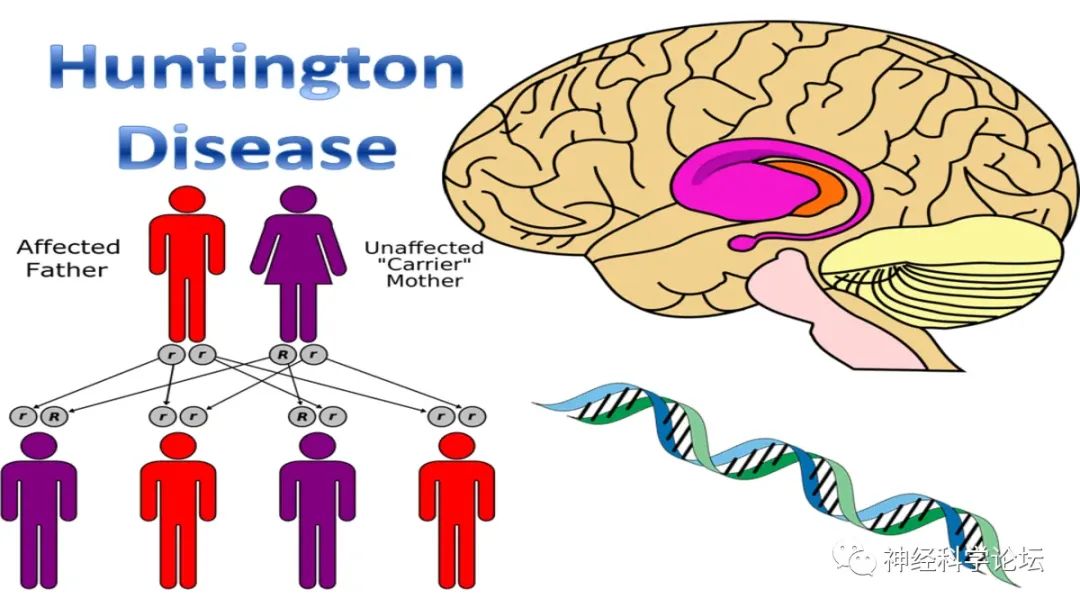
亨廷顿病是一种罕见的常染色体显性遗传性神经系统变性疾病,典型表现包括逐渐进展的舞蹈样动作、认知障碍和精神行为异常。亨廷顿病在各种族人群均有报道,以高加索人种多见,患病率为(10.6~13.7 ) /10万;亚洲地区罕见,日本、中国台湾和香港地区患病率约为(0.1~0.7 ) / 10万,中国大陆目前缺乏流行病学数据。欧洲人后裔的发病率是东亚人的10-100倍。根据1985年至2010年发表的研究,HD的总发病率估计为每100,000人年0.38人,全球总患病率为每100,000人年2.71人。最近的研究强调,在一些地区,流行率有所上升。尽管对不同人群中的差异和增加率有合理的解释,如不同HTT基因单倍型的识别、医疗保健的可及性、对疾病相关污名的不同态度、迁移和HD聚集区的识别,但具体的决定因素仍有待阐明。近10年来,该病的诊疗有了较大进展,尤其是在治疗方面,包括药物可及性和物理治疗的建议。为更好地规范和指导亨廷顿病的临床诊疗,本指南对亨廷顿病的临床表现、诊断、鉴别诊断、治疗及遗传咨询等方面进行了系统阐述和规范推荐,对现阶段亨廷顿病的临床诊疗有重要的指导意义。
临床表现
亨廷顿病平均发病年龄在35-45岁之间。患者男女发病无差别,这种情况通常持续16-18年。随着病情的发展,对日常工作的依赖性增加,最终导致死亡。亨廷顿病的初始运动指征可能是肌张力障碍。抽搐是另一种不受欢迎的运动,尽管它们相对不常见。小脑主诉可能表现不稳定,与近视和近视很相似。在许多情况下,运动被比作“醉酒”或经历小脑共济失调。区分舞蹈症和共济失调步态是非常具有挑战性的。不论发病年龄,HD都是一种慢性、缓慢进行性疾病。临床发病后的平均存活时间为10~20年,部分患者可存活30~40年。可在任何年龄起病,但多见于40~45岁,也有18个月和80多岁起病者。依据起病年龄的不同,将亨廷顿病患者分为青少年型(≤20岁)、成年型(21~59岁)和老年型(≥60岁)。
运动障碍
运动障碍主要分为不自主运动和自主运动障碍两类。其中以舞蹈样运动最常见,其次是肌张力障碍、帕金森样表现等。亨廷顿病患者早期主要表现为舞蹈样运动,随病程进展,部分会出现肌张力障碍和帕金森样表现等,最终丧失自主活动能力。
1.舞蹈样运动:多见于成年型患者以及早中期老年型患者。这是一种不受控制、无目的、无节律、运动幅度不一致的不自主运动。病程初期多为不易察觉的手、足、眼和口周部不自主动作,后逐渐出现容易察觉的异常动作,如挤眉弄眼、撅嘴、伸舌、转颈耸肩、伸屈手指弹钢琴样动作等,常伴有肌张力降低。
2.肌张力障碍:多见于青少年型患者以及中晚期成年型患者。面部受累出现表情怪异、扭曲、口下颌肌张力障碍;颈部受累出现扭转、后仰、侧倾或复合类型颈部异常姿势;躯干受累出现扭转痉挛表现。症状从轻微的间歇性异常运动伴或不伴姿势异常,到肢体躯干的严重扭转,对日常生活和工作产生严重影响。
3.帕金森样表现:多见于青少年型患者以及中晚期成年型患者。部分患者可出现肌张力增高伴运动迟缓。
4.共济失调:部分患者早期运动症状不典型,表现为共济失调,容易误诊和漏诊。
5.其他运动障碍:少数患者可能出现肌阵挛、静坐不能、磨牙等表现。晚期患者可因各种运动障碍导致吞咽困难、构音障碍、平衡障碍和步态异常,反复跌倒、无法独立行走,最终卧床,还可因反复肺部感染和重度营养不良,严重威胁生命。
认知障碍
亨廷顿病患者在整个病程中几乎均有不同程度的认知障碍,甚至在出现运动障碍前10年已有认知下降,主要以执行力和注意力损害为主。患者早期记忆力下降不明显,故不易被察觉,可通过Stroop测验、符号数字测验及语义流畅性测验等发现和评估亨廷顿病患者的认知损害,病情常进行性进展为痴呆。
精神行为异常
约33%~76%的亨廷顿病患者有不同程度的精神障碍,表现形式多样,常见抑郁、易激惹及淡漠,其他还包括躁狂、攻击性行为、强迫、焦虑、幻觉和妄想等。
非特异性症状
非特异性症状包括睡眠障碍、体重减轻、疼痛、多汗、唾液分泌过多、口腔健康状况不佳、肺功能下降、呼吸肌肌力下降、尿失禁及性功能障碍等,有时可以早于其他症状出现。
特殊类型
1.青少年型亨廷顿病:青少年型亨廷顿病是指起病年龄小于或等于20周岁的患者。其临床表现与成年型亨廷顿病不同,主要表现为肌张力障碍、抽动障碍、共济失调、肌阵挛、肌强直、运动迟缓及癫痫发作等,而舞蹈样动作在疾病早期并不常见。10岁以内发病者,语言发育迟缓较为突出。
2.老年型亨廷顿病:老年型亨廷顿病是指起病年龄大于或等于60岁的患者,起病症状比较单一,以舞蹈样动作为主,病程进展与成年型亨廷顿病无明显差异,但生存年限较短。衰老、恶性肿瘤、脑血管疾病或跌倒意外是导致死亡的主要原因。
临床分期
亨廷顿病分为症状前亨廷顿病和症状期亨廷顿病。
症状前亨廷顿病个体一般是先证者的无症状同胞或子女,其HTT基因检测提示异常,但未出现明显临床症状,可分为2期:
(1)无症状期:此期亨廷顿病个体与健康个体无法区分;
(2)前驱期:此期亨廷顿病个体的运动、认知和行为发生细微改变,统一亨廷顿病评定量表(UHDRS)的运动症状评分小于5分。
症状期亨廷顿病临床上可分为3期:
(1)早期:症状不明显,可有轻微的不自主运动、行动笨拙、眼球运动异常和嗅觉异常,可出现抑郁、焦虑、淡漠或易激惹、注意力和执行能力下降,但患者仍可工作,并具有独立生活能力;
(2)中期:出现明显的运动障碍,以舞蹈样动作为主,动作不协调、易跌倒,还可出现肌张力障碍、运动迟缓、言语异常和体重减轻等表现,认知功能进一步减退,无法处理复杂事务,但日常生活能力基本保留;
(3)晚期:患者丧失日常生活能力,多数卧床不起,需要他人照顾,舞蹈样动作可能加重,但常被肌强直或肌张力障碍掩盖,并可出现言语不能、体重明显减轻以及因吞咽障碍导致误吸和窒息等表现。
辅助检查
量表评估
可采用UHDRS评估疾病严重程度,评分越高,提示病情越严重。
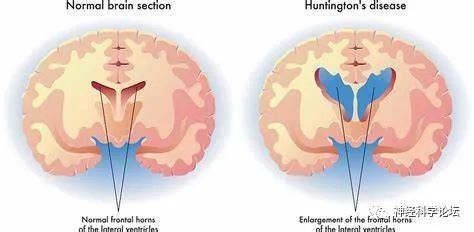
影像学检查
典型患者颅脑MRI和CT可发现大脑皮质、尾状核及壳核萎缩,以尾状核头部最为显著,还可见侧脑室前角扩大。早期患者可无异常发现。高分辨率和功能磁共振发现症状前亨廷顿病个体已出现尾状核萎缩,至临床症状明显时,体积已缩小过半,且临床症状出现前10年已出现脑萎缩和神经纤维传导束受损。
生物标志物检测
血清神经丝轻链蛋白(NfL)可用于评估亨廷顿病患者的起病年龄和疾病进展程度。相较于血清NfL,脑脊液NfL水平可以更敏感地预测亨廷顿病的疾病进展。
HTT基因检测
一般采用Sanger测序或毛细管电泳技术检测HTT基因1号外显子上的CAG重复次数,前者对CAG重复次数判定更为精确。不推荐采用第二代和第三代测序技术常规筛查HTT基因。正常人CAG重复次数≤26;当CAG重复次数为27~35时,不会发病但传递给子代时可出现CAG重复次数扩增,子代可能因此发病;当CAG重复次数为36~39时,不完全外显,部分携带者可不发病;当CAG重复次数≥40时,完全外显,所有携带者均会发病。
推荐意见:
(1)对临床疑诊亨廷顿病患者应行HTT基因检测(Ⅰ级推荐,B级证据);
(2)为明确HTT基因的CAG重复次数,建议优先使用Sanger测序技术进行检测(Ⅱ级推荐,C级证据);
(3)血清和脑脊液NfL水平可用于评估亨廷顿病患者的起病年龄和疾病进展,脑脊液NfL更为敏感(Ⅰ级推荐,A级证据)。
诊断标准
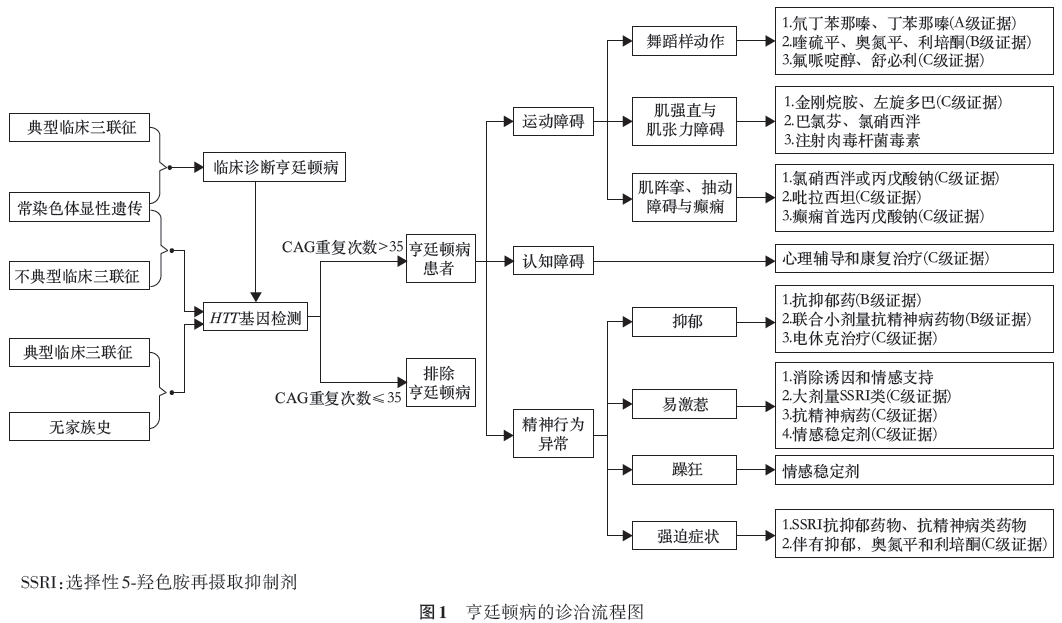
1.典型临床三联征:逐渐进展的舞蹈样动作、认知障碍和精神行为异常;
2.常染色体显性遗传家族史;
3.HTT基因检测结果:CAG重复次数大于35次。
推荐意见:
(1)符合1和2,临床诊断亨廷顿病,进一步检测HTT基因,符合3则基因诊断确诊为亨廷顿病(Ⅰ级推荐,A级证据)
(2)若未见典型临床三联征,但具有常染色体显性遗传家族史,建议行HTT基因检测,以助排除亨廷顿病诊断(Ⅰ级推荐,C级证据);
(3)仅符合3,则为症状前亨廷顿病。
鉴别诊断
神经棘红细胞增多症
常染色体隐性或X连锁遗传,致病基因主要有VPS13A、XK和PANK2。临床表现与亨廷顿病类似,也表现为运动障碍、认知障碍和精神行为异常。运动障碍包括舞蹈样动作和肌张力障碍,以口面部肌张力障碍最常见。与亨廷顿病不同之处在于,50%患者可伴有癫痫和(或)周围神经病、远端肌萎缩和血清肌酶增高,且外周血涂片可见棘红细胞。
遗传性共济失调
脊髓小脑性共济失调(SCA)1、2、3、17型及齿状核‑红核‑苍白球变性(DRPLA)呈常染色体显性遗传,临床上常出现共济失调、震颤、肌阵挛、癫痫、舞蹈样动作等表现,表型复杂。
SCA1、2和3型患者,主要以进行性加重的小脑性共济失调为核心表现,典型患者易与亨廷顿病鉴别,但需注意少数患者早期可出现不自主运动而无共济失调表现,临床医生应注意进行鉴别。
SCA17与亨廷顿病临床表现较为相似,若无共济失调症状时,临床上常难以鉴别,但此型患者较为罕见,国内仅见零散报道。
DRPLA可出现舞蹈样动作和认知障碍,但共济失调表现和肌阵挛癫痫易与亨廷顿病区别,且头颅影像易出现白质高信号。
良性遗传性舞蹈病
多数患者在婴儿期发病,出现舞蹈样动作,运动发育迟缓,行为异常,肌张力障碍,甚至肌阵挛性抽动。现已报道ADCY5、NKX2‑1、PDE10A、GNA01和SLC16A2与该病表型相关。
类亨廷顿病综合征(HDL)
患者有舞蹈样动作、人格改变和认知障碍等表现,易与亨廷顿病混淆。
HDL1起病年龄偏早,进展缓慢;HDL2几乎只见于非洲裔人群;HDL3目前仅在沙特阿拉伯家系中发现,极为罕见,且为常染色体隐性遗传模式。HDL确诊需依靠基因检测。
PRNP基因突变与HDL1表型相关,而JPH3基因的CTG三核苷酸重复扩增超过39次时可确诊HDL2。
获得性舞蹈症
常见者如小舞蹈病,又称风湿性舞蹈病,是风湿热在神经系统的表现,常见于儿童和青少年,患者多有风湿热或链球菌感染病史,亚急性或急性起病,自限性病程,其临床特征为舞蹈样动作、肌张力降低、肌力减弱及精神症状等。此外,还需仔细甄别糖尿病非酮症性偏侧舞蹈症,长期使用抗精神病药物诱发舞蹈症,以及血管性、感染性和免疫相关性舞蹈样病变等。
临床治疗
目前尚无延缓亨廷顿病病程进展的疾病修饰药物,故本病强调综合性治疗,包括药物对症治疗、协同康复及心理治疗,在疾病的不同阶段各有侧重。药物对症治疗的主要目标是控制症状,提高生活质量。由于亨廷顿病的症状随病程进展而变化,故需规范定期随访,适时调整用药方案。多数药物有不良反应,应从小剂量起始给药,滴定增量,尽量避免多药联合。饮食干预主要为地中海饮食,如大量食用蔬菜、水果、豆类、鱼类等,可改善患者的认知功能、运动能力和生活质量。
推荐意见:
(1)对亨廷顿病患者的舞蹈样动作,优先推荐丁苯那嗪或氘丁苯那嗪治疗,服药期间注意抑郁表现、肝功能异常及QT延长等不良反应(Ⅰ级推荐,B级证据);考虑药物可及性和经济负担等因素,抗精神病类药物仍可单用或联合氘丁苯那嗪改善舞蹈样表现(Ⅱ级推荐,C级证据)。
(2)亨廷顿病患者的认知障碍目前无有效的治疗药物,仍以心理辅导和认知训练为主(Ⅱ级推荐,C级证据)。
(3)精神行为异常的治疗药物选择因个体表现不同,针对性选择不良反应较少的抗精神病药物及改善心境药物(Ⅱ级推荐,B级证据)。
(4)规范的物理及认知康复治疗,有利于患者运动和认知功能的改善(Ⅱ级推荐,A级证据)。
小编未来展望:专注于理解导致HTT基因突变的潜在分子机制的研究非常有希望,旨在找到治疗HD的方法。然而,目前对HD的治疗仍然有限。所应用的治疗集中在症状的治疗上,因为防止疾病发作和减缓疾病发展的神经保护疗法还不可用。近年来,随着疾病的发展,对细胞病理学和大脑总体结构变化的研究取得了巨大进展。越来越多的证据支持炎症在神经退行性疾病中起着关键作用,并刺激了免疫治疗策略在调节神经炎症性疾病中的应用。一些潜在免疫调节药物的临床前和临床试验已在HD中进行,如米诺环素和大麻素类药物、拉喹莫德、TNF-α抑制剂、抗SEMA4D单克隆抗体、神经节苷脂等。HD进展期可持续10年或更长时间,取决于治疗/护理水平。不动的并发症(例如,吸入性肺炎和其他感染)导致患者在发病后10-40年死亡。
原文索引:中华医学会神经病学分会神经遗传学组.中国亨廷顿病诊治指南2023.中华神经科杂志,2023,56(8):848-855.

