Nat Metab 北京大学肖武生揭示血管平滑肌细胞中HIF1α常氧激活的分子机制
时间:2024-11-08 06:02:12 热度:37.1℃ 作者:网络
缺氧诱导因子1α (Hypoxia inducible factor 1α, HIF1α)是调控低氧环境下生物过程的主要因子。在正常氧条件下,HIF1α蛋白被脯氨酸羟化酶(Prolyl hydroxylase, PHD)羟基化,随后被E3泛素连接酶复合体von-Hippel Lindau蛋白(pVHL)识别,导致多泛素化和蛋白酶体降解。因此,PHD酶的失活可促进HIF1α蛋白稳定和下游靶点激活,从而调节众多生理和病理过程。已有研究发现,常氧环境下,肿瘤细胞的HIF1α通路可被激活。然而,在正常原代细胞中,HIF1α信号的常氧激活及其潜在机制仍知之甚少。
2024年10月29日,北京大学公共卫生学院肖武生助理教授与哈佛医学院和布莱根妇女医院Joseph Loscalzo教授团队在Nature Metabolism杂志发表题为“Branched-chain α-ketoacids aerobically activate HIF1α signalling in vascular cells”的研究论文。该研究揭示了支链氨基酸的α-酮酸衍生物(Branched-chain α-ketoacids, BCKAs)是尚未被识别的信号代谢产物,可诱导HIF1α常氧激活;该BCKA-HIF1α途径参与调节血管平滑肌细胞表型与功能,且与肺动脉高压(Pulmonary arterial hypertension, PAH)病理相关。

鉴于HIF1α是血管生成、血管功能稳态以及血管相关疾病的关键调节因子,且调控HIF1α活性是治疗心肺血管疾病的潜在策略,本文研究了正常原代血管细胞,尤其是血管平滑肌细胞(VSMCs)中HIF1α常氧激活的可能机制及其与肺动脉高压的发生关联。
研究人员首先检测了人原代血管细胞和成纤维细胞以及肿瘤细胞常氧培养下的HIF1α蛋白水平,发现人肺动脉平滑肌细胞(Pulmonary artery smooth muscle cells, PASMCs)和主动脉平滑肌细胞表达HIF1α蛋白,但其在相同培养条件下的血管内皮细胞、肺成纤维细胞、真皮成纤维细胞、肺和乳腺癌细胞中不表达。VSMCs中有氧HIF1α稳定与较高的糖酵解相关靶基因mRNA表达水平和乳酸含量相关,且高HIF1α基础活性抑制了缺氧诱导的糖酵解活性进一步增强。
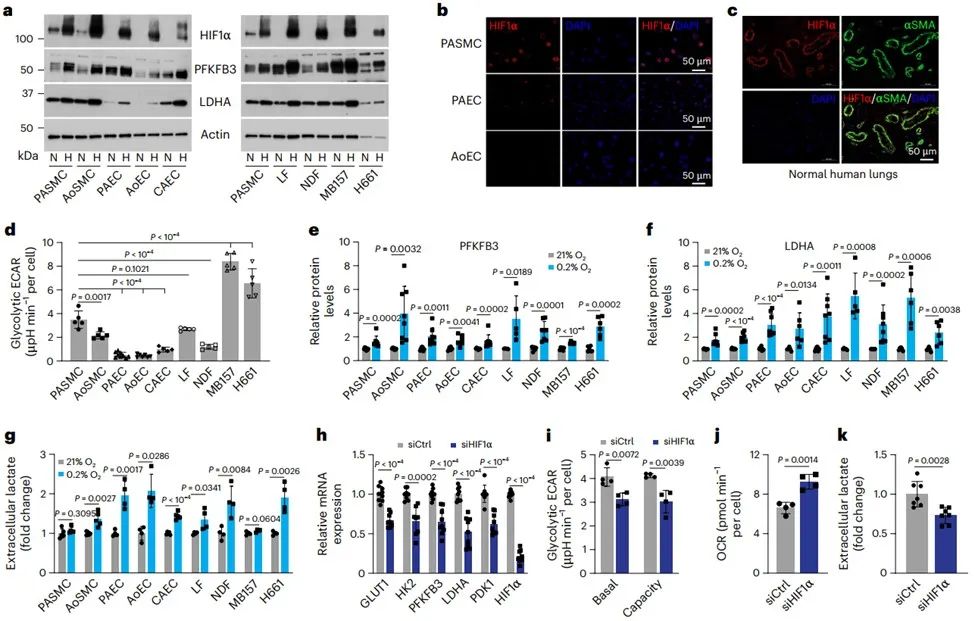
通过条件培养基(Conditioned medium, CM)进行PASMCs传代培养,发现与在常规生长培养基中培养的细胞相比,CM培养的PASMCs中HIF1α蛋白及其靶基因表达水平更高(CM选择性和特异性诱导HIF1α而非HIF2α蛋白),且与PHD2酶活性被抑制相关。CM培养的PASMC中糖酵解活性增强依赖于HIF1α。因此,推测常氧条件下PASMCs中HIF1α蛋白的稳定和激活可能是由旁分泌分子介导的。
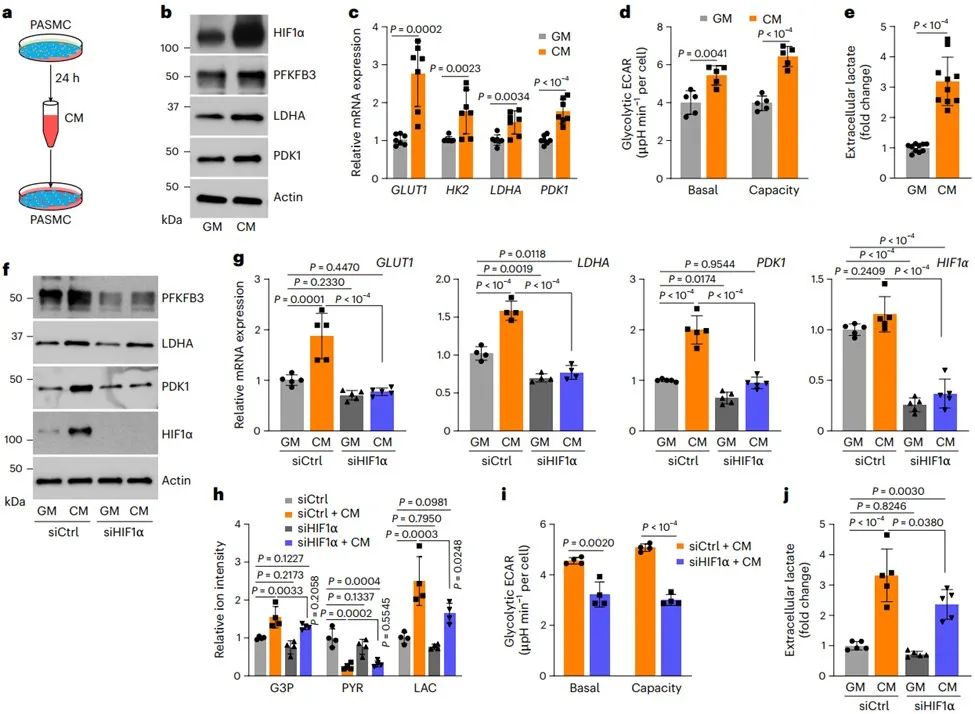
接下来,研究人员确定了旁分泌分子可能为小分子物质,并通过代谢组学分析筛选出3种代谢物KIC、KMV和KIV(分别是支链氨基酸亮氨酸、异亮氨酸和缬氨酸的代谢产物)。处理与CM中相当浓度的BCKAs足以上调HIF1α蛋白及其应答基因表达,且单独敲低支链氨基酸转移酶2 (Branched-chain amino acid transaminase, BCAT2)或BCAT1/2双沉默的PASMCs于CM中培养,HIF1α信号显著激活,证明BCAT2催化产生的BCKAs(特别是KIV)是有氧HIF1α激活的旁分泌介质。
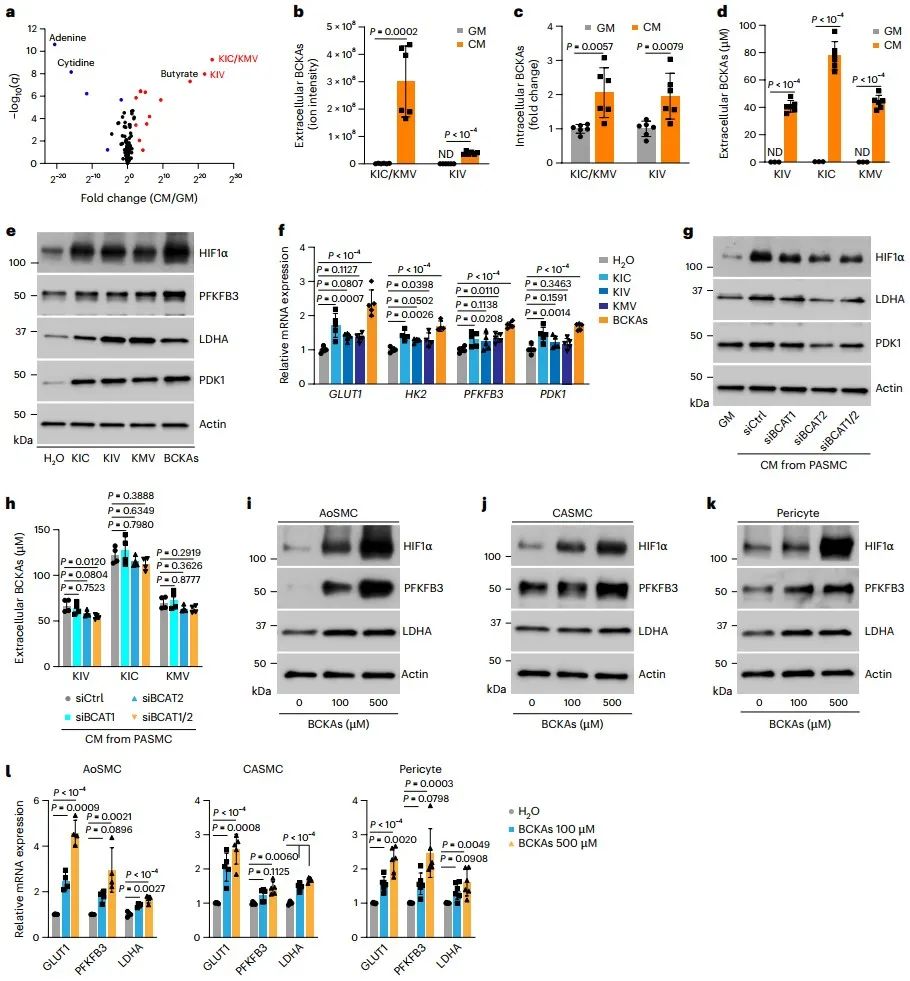
BCKAs介导的有氧HIF1α激活的机制是什么?分子对接和计算机模拟预测分析显示,BCKAs可与PHD2酶的催化活性中心Fe2+结合,表明BCKAs可能是铁螯合剂,从而抑制PHD2活性并稳定HIF1α蛋白。生化实验证实BCKAs可直接抑制PHD2酶活性,IC50为252 μM,高于稳定体外细胞HIF1α的浓度(50-100 μM),提示可能存在其他抑制机制。进一步的研究发现,BCAT1将BCKAs转化为BCAAs和α-KG,后者在乳酸脱氢酶A (Lactate dehydrogenase A, LDHA)催化下生成L-2-羟基戊二酸(L-2-hydroxyglutarate, L2HG;一种已知的PHD2酶强抑制剂)。通过对LDHA和L-2-羟基戊二酸脱氢酶(L2HG dehydrogenase, L2HGDH)表达以及L2HG水平的调控,进一步证明LDHA介导的L2HG抑制PHD2活性,从而导致HIF1α信号的有氧激活。不完全同于缺氧激活HIF1α诱导的代谢重编程,BCKAs介导的有氧HIF1α激活增加了糖酵解和三羧酸循环相关代谢物的水平,提示糖酵解和氧化磷酸化活性均增强。
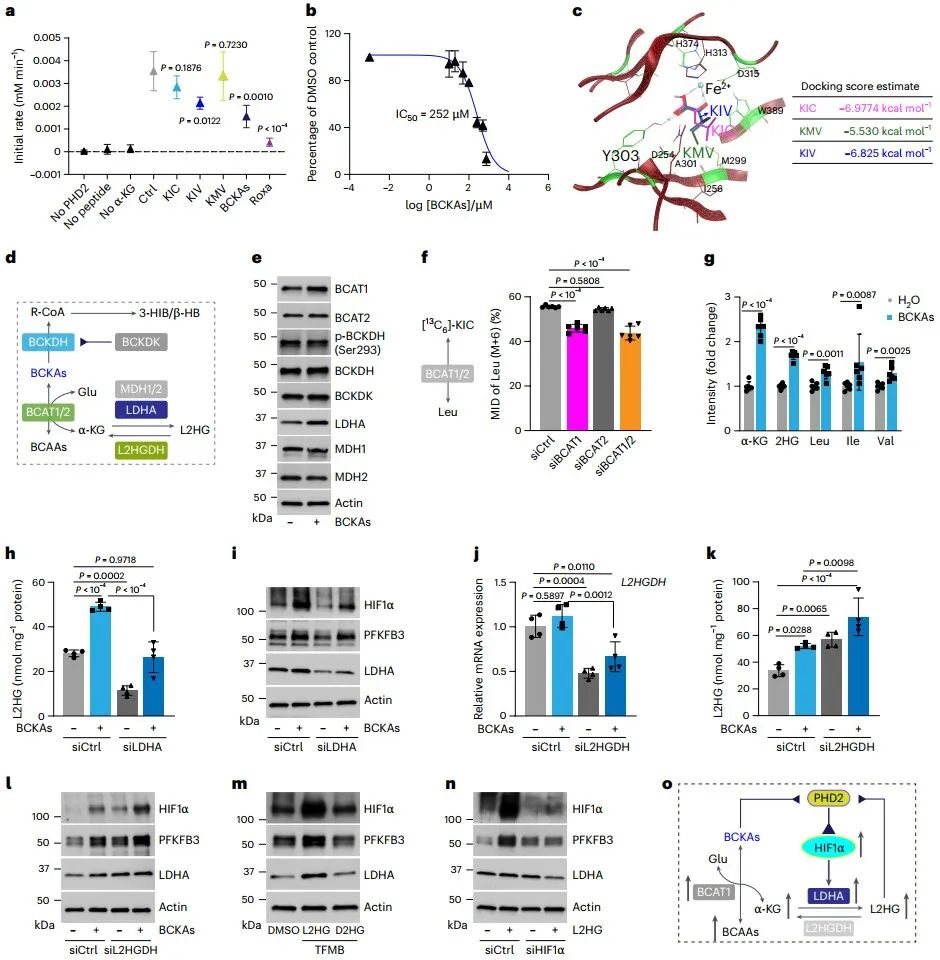
最后,研究人员对上述通路机制与VSMCs表型以及PAH病理的关联进行了研究,发现BCKAs能促进PASMCs由收缩亚型向合成亚型转换,表现为上调了合成亚型标志蛋白VIM的表达和增强了I和IV胶原蛋白的合成与分泌。有趣的是,HIF1α激活可能在该效应中发挥负反馈调控,因为敲低HIF1α反而进一步增强了BCKAs诱导的VIM和胶原蛋白的表达。PAH是一种严重的心肺血管疾病,其特征是HIF1α激活,糖酵解增强,胶原沉积和纤维化增加以及PASMC功能障碍。研究人员发现,特发性PAH (Idiopathic PAH, IPAH)患者肺组织中BCKAs关键代谢基因BCAT2、支链酮酸脱氢酶激酶(Branched-chain ketoacid dehydrogenase kinase, BCKDK)和支链酮酸脱氢酶E1亚基α (Branched-chain ketoacid dehydrogenase catalytic E1 subunit α, BCKDHA1)表达上调,而BCAT1表达下调,与KIC和KMV水平显著升高以及HIF1α蛋白增加相关。类似的结果在3种PAH模型雄性大鼠中也观察到。此外,利用IPAH患者来源的PASMCs (IPAH-PASMCs)细胞,进一步的研究发现,PASMCs主导了PAH肺中BCKAs代谢基因的变化、BCKAs水平累积及其诱导的代谢和表型改变。因此,PAH模型中BCKAs代谢失调导致细胞内BCKAs积累,并随之促进IPAH-PASMCs中糖酵解活性增强和合成表型,从而参与PAH的发生和发展。
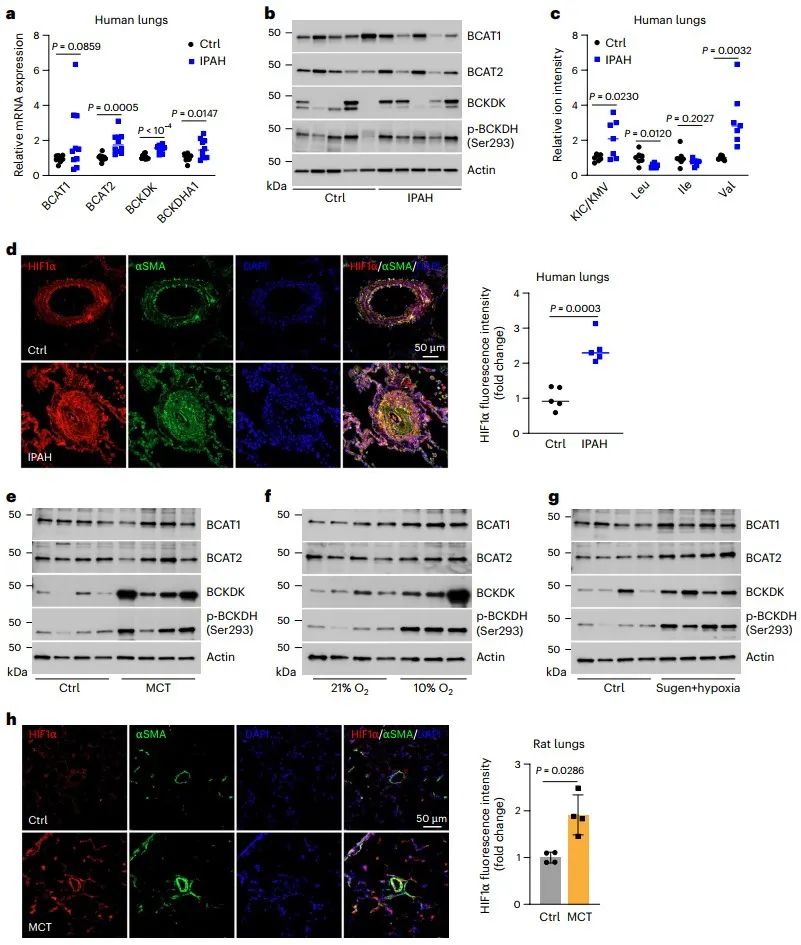
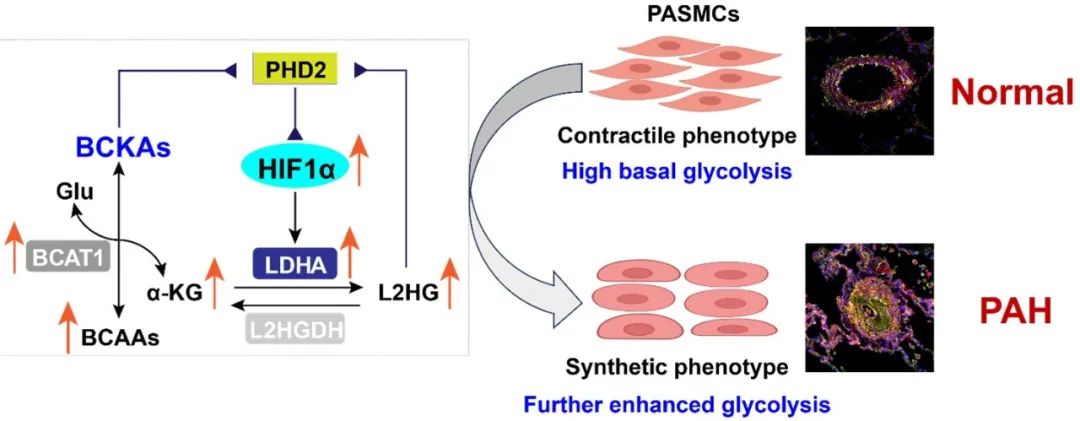
综上所述,BCKAs是尚未被识别的信号代谢产物,可通过直接和间接的双重机制来抑制PHD2活性和激活HIF1α信号,进而增强糖酵解活性,调控PASMCs的合成表型转化,最终参与PAH等肺血管功能障碍和疾病发生。
原文链接:
https://www.nature.com/articles/s42255-024-01150-4

