知识宝典!27种引起脑室扩大的病变
时间:2021-10-29 06:02:15 热度:37.1℃ 作者:网络
一、侧脑室局部扩大脑室局部扩大 1.局限性脑萎缩
外伤后脑萎缩,感染后脑萎缩,脑梗死后脑萎缩CT可见扩大的脑室部分附近的脑实质呈片状低密度灶MR:T1WI呈低信号,T2WI呈高信号。同时脑沟及蛛网膜下腔增宽。
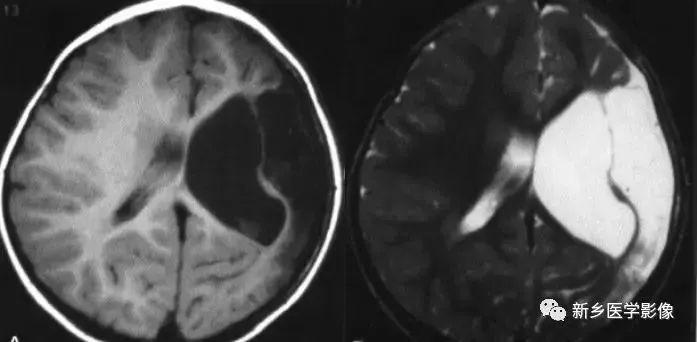
2.侧脑室神经上皮囊肿
通常位于侧脑室三角区,囊壁薄,通常显示不清由于囊内含脑脊液,所以CT及MR都表现为脑脊液信号。
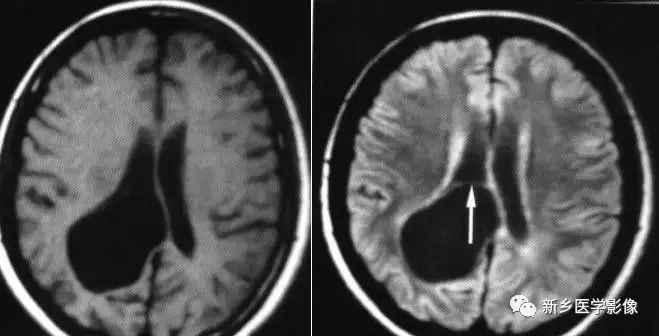
3.解剖变异
特点为附近脑实质内无病变存在,无脑沟及蛛网膜下腔扩大或脑结构异常
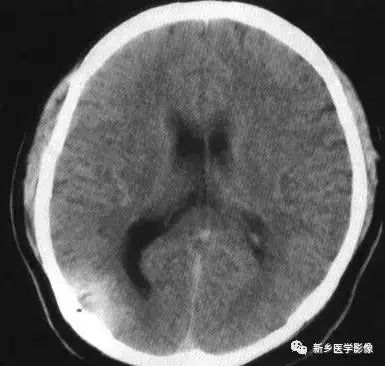
4.孤立性侧脑室颞(下)角
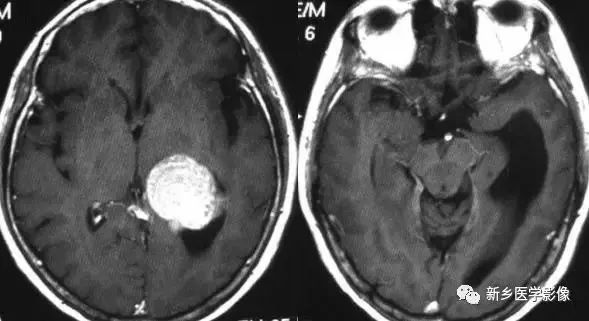
是侧脑颞角的脑脊液流动受阻所致.常因侧脑室病变所致,包括室管膜内出血、脑膜瘤和脉络膜从乳头状瘤等,也可为侧脑室三角区周围病变压迫所致。
二、一侧脑室扩大
1.正常变异
正常人中,有相当一部分双侧侧脑室大小不一致.不对称一侧侧脑室明显大一于另一侧。临床上,智力及精神发育正常,无临床意义。
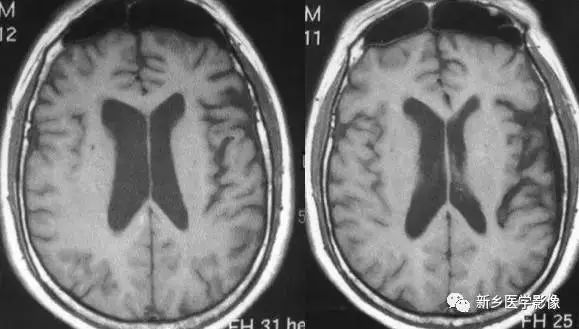
2.一侧大脑半球萎缩
可由许多原因引起,如脑梗死、外伤、出血及感染等。最常见的原因是血管闭塞引起大面积脑。CT和MRI现为患侧侧脑室扩大、脑组织量减少,中线向患侧移位,严重者脑沟和脑回消失不见。
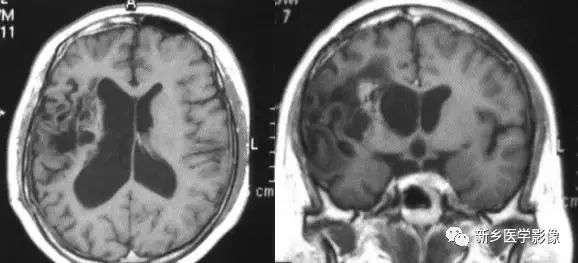
3.脑三叉神经血管瘤病
又称为脑颜面血管瘤综合征,CT平扫以一侧半球脑实质内脑回样、轨道样、弧带状或锯齿状钙化为特征。由于受累侧血供障碍,常引起脑实质萎缩.所以可以表现有患侧侧脑室扩大,颅腔变小,颅板增厚。脑内典型钙化及临床三叉伸经分布区有紫红色血管痣。
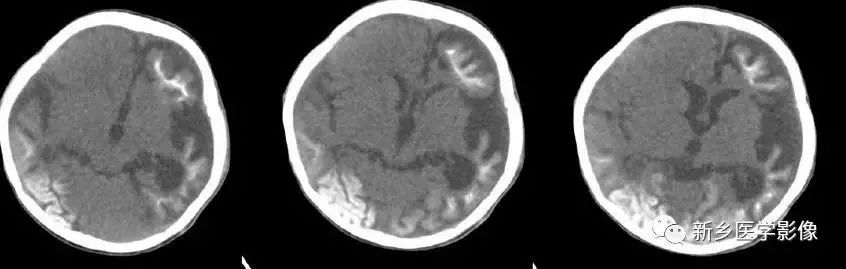
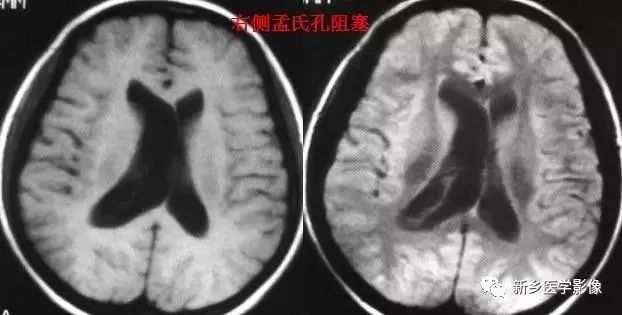
4.一侧室间孔阻塞
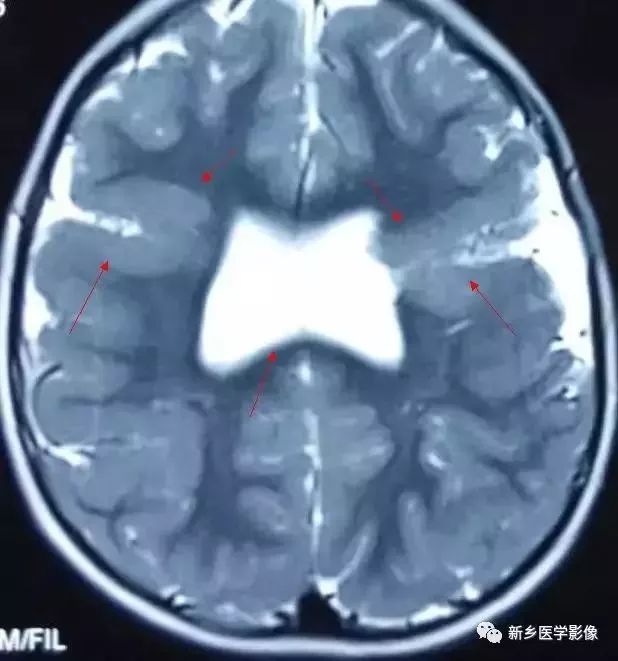
脑脊液主要产生于脑室的脉络膜丛。当一侧室间孔阻塞时,同侧侧脑室脉络膜丛产生的脑脊液不能进入三脑室,脑脊液在侧脑室内聚积,CT和MR检查表现为一侧侧脑室扩大,扩大明显时可有中线结构向对侧移位,主要原因有室间孔附近的肿瘤、囊肿、囊虫及炎性粘连。确定一侧侧脑室扩大为室间孔阻塞所致的要点包括:一侧侧脑室扩张明显,有张力.透明隔向对侧移位;扩一大的侧脑室周围没有能够导致该侧侧脑室扩大的其他可以解释的原因。
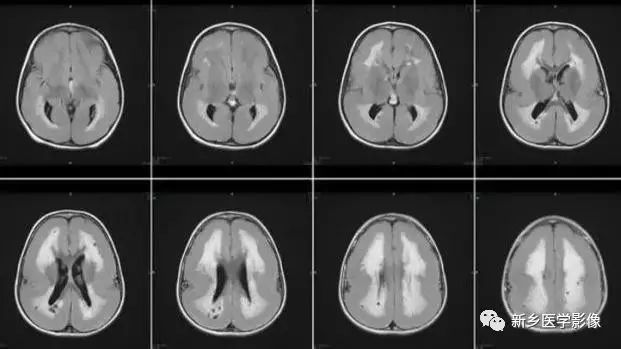
5.脑室周围白质软化症
主要与缺血缺氧及感染有关.常见于早产儿.是造成早产儿脑瘫的主要原因。由于脑室周围白质的血供分别来自脑室区和远脑室区的终动脉。未成熟儿终动脉深穿支的侧支循环尚未建立,而胚胎晚期脑室周围白质对缺血缺氧敏感。所以,脑室周围自质软化症多见于早产儿。由于病灶常为双侧性,故双侧侧脑室多同时扩大。脑白质内软化灶在CT扫描时表现为白质内斑片状低密度灶.MR T1加权图呈低信号,T2加权图呈高信号。
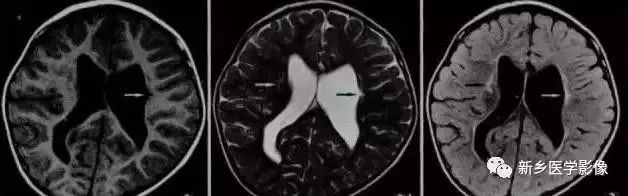
三、双侧侧脑室扩大
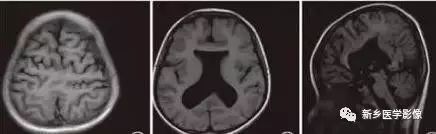
1.普遍性脑萎缩
普遍性脑萎缩常同时累及灰质和白质.可表现有双侧侧脑室扩大。而且是双侧侧脑室轻度对称性扩大最常见的原因。尤其是以脑白质萎缩为主的病人。
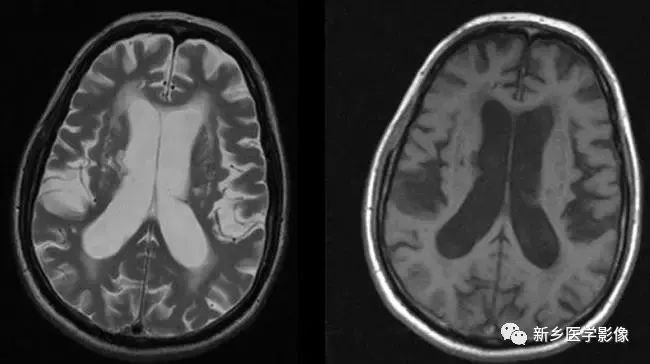
普遍性脑萎缩引起的侧脑室扩大通常比较对称.中线无移位。其特点是同时有脑沟、脑裂增宽等脑皮质萎缩的表现.
普遍性脑萎缩可见于正常老年人
原因有:①与年龄有关的脑萎缩CT和MR检查可见脑室、脑池轻度扩大.脑沟轻度增宽.常以额叶和镰旁更明显。②阿尔茨海默病本病除具有普遍性脑萎缩的一般表现外.颞叶萎缩常很明显且现较早,故侧脑室扩大以颞角更明显。阿尔茨海默病神经元丧失的严重程度从重到轻依次为海马、颞叶后部、额叶和顶叶。③Huntington‘s病(舞蹈症)是一种常染色体显性遗传的基底节和大脑皮层变性性疾病。主要损害基底节和大脑皮层,尾状核、壳核病变最明显。小神经节细胞严重破坏,大细胞也减少、尼氏体消失、核固缩、出现类淀粉小体,还有脱髓鞘改变和胶质增生,基底节部受累常最明显且发生最早。临床主要根据3大特征:舞蹈样动作、痴呆、家族史。④帕金森病又称震颤性,是一种常见的锥体外系疾病。临床以震颤、肌强直和运动障碍特征。震颤喂首发症状,休息和安静时明显。CT除有萎缩外,有时可见基底节钙化。MR T2WI上基底节区和脑白质内常有多发高信号斑点存在⑤匹克病(Pick’s disease)又称脑叶硬化症女性多见,50岁为发病高峰,临床以进行性痴呆为主要表现。CT和MR常以额叶和颞叶萎缩为主。另一特点,双侧萎缩常不一致,左侧较明显,颞上回的前半部萎缩,而后部常正常。⑥Jakob-Creutzfeldt’s病一种以迅速进行性痴呆为特点的脑病CT和MR表现为侧脑室对称性扩大,脑沟脑裂增宽。短期复查可见萎缩程度明显加重,晚期可出现脑白质弥漫性脱髓鞘改变。⑦其他原因如缺氧、中毒、物理损伤、营养不良等。
2.早产儿侧脑室扩大
在胚胎早期,侧脑室相对很大,随着发育的不断成熟.侧脑室逐渐变小.到胚胎36周时达正常大小,故早产儿常表现有双侧侧脑室轻度对称性扩大,龙其是30周前出生的早产儿。可以同时合并有或不伴有早产儿颅内出血。
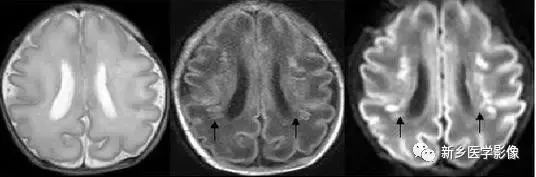
3.巨脑畸形
巨脑畸形可同时表现有双侧侧脑室对称性扩大.通常呈轻度。这种巨脑畸形可以是许多疾病的伴发征象:如亚历山大病、海绵状变性、Soto's综合征等。常见的情况是,许多大头儿童临床智力发育及体格发育正常,无颅压增高表现.无神经系统体征及症状。
Soto's综合征又称脑性巨人症,在新生儿期即表现有身体发一育显著增快.并有大头巨脑畸形、神精发一育迟缓、而容特别、前额突出、睑裂向外下倾斜.眼距i过宽、下颌细而突出,生长激素及内分泌检查正常,头颅CT和MR检查时主要表现为巨脑及侧脑室扩大。诊断主要要结合上述临床其他情况。
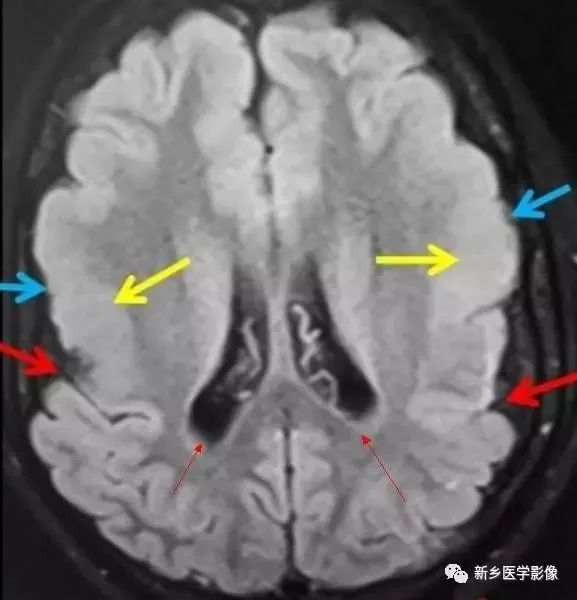
4.大脑先天发育异常
许多种大脑发育异常均可同时表现又侧脑室扩大,包括前脑无脑裂畸形、脑裂畸形、无脑回和巨脑回等。
(1)前脑无裂畸形: (holoprosencephaly)是指一系列位于中线程度不同的畸形,累及大脑、面部、脑于和小脑.前脑未能够分开,呈不全性或完全性,端脑和间脑无法区分。根据脑及面部畸形程度将其分为无叶型、半叶型和单叶型。
无叶型前脑无裂畸形最严重.端脑半球间没有裂隙,呈马蹄形或新月形扩大的单脑室跨越中线,与背侧囊交通。丘脑互相融合,面部畸形从两眼距离过近到独眼畸形.往往死于胎儿或新生儿期。
半叶型前脑无裂畸形在前脑可见部分裂隙. 形成不同发育程度的大脑纵裂及大脑镰。两侧大脑半球在前部未完全分开,但枕叶和 双侧侧脑室体部分离,丘脑分开不完全。三脑室和海马发育不全,胼胝体仅可见到压部,而其他部分缺如.额叶和基底节前部分辨不清。临床表现有两眼距离过近.及唇裂、腭裂等面部畸形,侧脑室呈单一性.且明显扩大。
单叶型前脑无裂畸形与正常发育脑仅有些很小的区别.如透明隔缺如或双侧额叶不完全分开。
(2)脑裂畸形:胚胎期脑的发育经历6个主要阶段:①背侧诱导阶段;③腹侧诱导阶段;③神经增生阶段;④神经元移行阶段;⑤组织形成阶段;⑥髓鞘形成阶段。脑裂畸形发生在神经元移行阶段。脑裂畸形可累及一侧或双侧大脑半球,脑裂畸形位于侧面.常累及中央前、后回区偶尔位于大脑半球的其他部位。脑裂畸形的裂隙可以很窄,裂隙两侧灰质紧密相贴,称闭合型。裂隙也可以很宽,中间为脑脊液,分离型。
分离型脑裂畸形需要与脑穿通畸形囊肿鉴别.脑裂畸形的裂隙两旁一定为一灰质结构,而脑穿通畸形囊肿周围无脑灰质包绕。裂隙两旁是否为灰质结构是区别脑裂畸形与脑穿通畸形囊肿的可靠征象。裂隙两侧的灰质可不正常,可呈多小脑回样。脑裂畸形也可合并脑灰质异位。
分离型在CT很容易显示.闭合型有时容易漏诊.MRI对裂隙两侧的灰质结构容易辨认。脑裂畸形常合并透明隔缺如.侧脑室扩大,脑裂畸形处脑室边缘不规则.常可见指向裂隙的裂或 三角形憩室存在。
临床上脑裂畸形常表现有癫痫发作,其他神经系统症状可从很轻微到很严重.主要取决于脑裂畸形使脑组织缺损的严重程度。单侧闭合型脑裂畸形症状通常较轻,双侧分离型脑裂畸形症状较明显。
(3)无脑回和巨脑回
无脑回和巨脑回是一组因神经元移行异常所致的脑回发育异常。巨脑回也称平滑脑。巨脑回指有部分脑回存在,这些脑回异常增大增宽.脑沟变浅。巨脑回主要位于额、颞部。无脑回上要位于顶、枕部。
临床上,无脑回和巨脑回畸形患儿均表现有小头畸形和轻微的面部异常,完全无脑回畸形常在两岁前死亡.不完全无脑回畸形存活常能长期。
CT和MR均能够很好显示无脑回和巨脑回畸,表现为大脑半球表面几乎呈光滑状,仅可见少数宽阔、粗大、平坦的脑回,脑沟缺如.脑灰质增厚,脑白质变薄,灰白质分界面异常平滑,见不到白质向灰质内伸入的现象。常见透明中隔腔存在.侧脑室扩大,蛛网膜下腔增宽。
5.双侧室间孔阻塞
与一侧室间孔阻塞一样,双侧室间孔阻塞后,可表现为双侧侧脑室对称性或不对称性扩大,脑室扩大通常很显著,脑室周围多伴有间质性脑水肿,双侧室间孔阻塞的原因与一侧室间孔相同。可同时伴有导水管狭窄。
诊断要点:双侧侧脑室扩大很显著而三脑室大小正常。
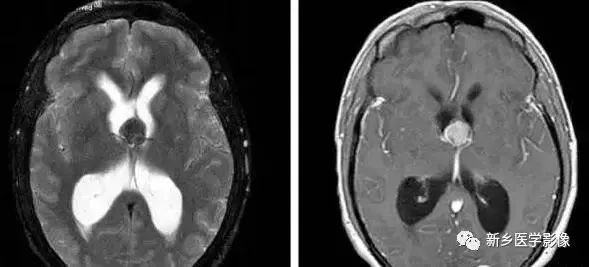
6.脑室周围白质软化症
主要与缺血缺氧及感染有关。
常见于早产儿。常见于早产儿的原因与胚胎期脑部损害发生的时间有关,胚胎早中期脑损害主要引发发育畸形,晚期主要引起脑血管改变。
由于脑室周围白质的血供分别来自脑室区和远脑室区的终动脉,未成熟儿终动脉深穿支的侧支循环尚未建立,而胚胎晚期脑室周围白质对缺血缺氧敏感。所以,脑室周围白质软化症多见于早产儿。
由于侧脑室周围有软化萎缩,故扩大的侧脑室外缘常不规则,不光整,这种不规则、不光整是本病引起脑室扩大的特征,另外,本病均表现有脑白质量减少及脑白质内斑片状软化病灶,脑白质减少严重时表现为部分区域白质消失,脑皮层与脑室侧缘接近甚至相连。CT表现为斑片状低密度灶。MR T1WI上呈低信号,T2WI呈高信号。
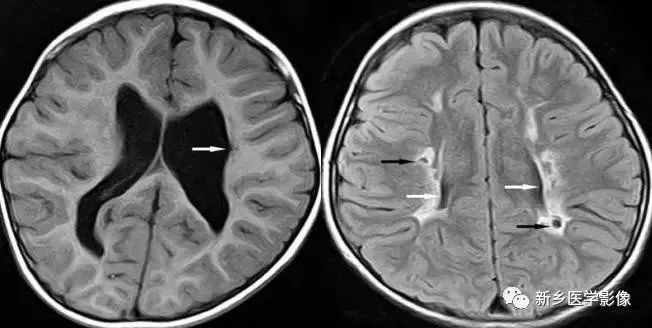
7.视-隔发育不良
是一种少见的先天发育畸形,主要包括:1.透明中隔缺如或发育不全,2.视神经、视交叉及漏斗部发育不良3.不同程度的下丘脑-垂体功能障碍。影像学表现可能很轻微,包括透明中隔缺如,视神经及视交叉变细,视神经管小,三脑室视隐窝扩大,双侧侧脑室扩大,侧脑室前角呈方形。多见于女性,可表现有尿崩症,视力障碍及丘脑下功能障碍。
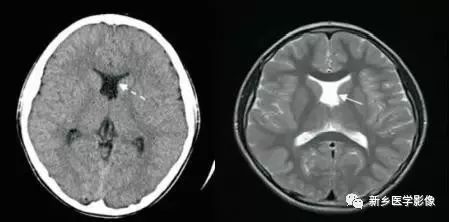
8.胼胝体发育不良
在脑发育腹侧诱导晚期,新形成的端脑嘴侧壁的背侧部分增厚、内陷,向后沿尚未完全发育的大脑半球间裂延伸。两个月后形成一个连合即胼胝体纤维的细胞框架,该细胞框架形成后胼胝体相应部分立即发育。膝部先发育。然后是体部、压部.位于胼胝体膝部后下方的胼胝体嘴最后发育。如果胼胝体发育过程中出现有害因素,就有可能导致胼胝体发育不良.表现为完全缺如或部分缺失。常表现为膝部存在或膝部和体部存在。压部和胼胝体嘴缺失。
胼胝体发育不良可见单独发病,但更常见的是伴有中枢伸经系统的其他畸形,包括胼胝体周围脂肪瘤、脑膨出、交通性脑积水Chaiarii畸形、Dandy-Walker囊肿、脑裂畸形等。临床上可无症状或仅有轻度临床症状,临床检查可见眼距过宽、大头畸形、智力发育迟滞等。胼胝体缺失时,MR冠状位上侧脑室前角呈新月形表现,侧脑室体部分离,呈垂直状平行走行。
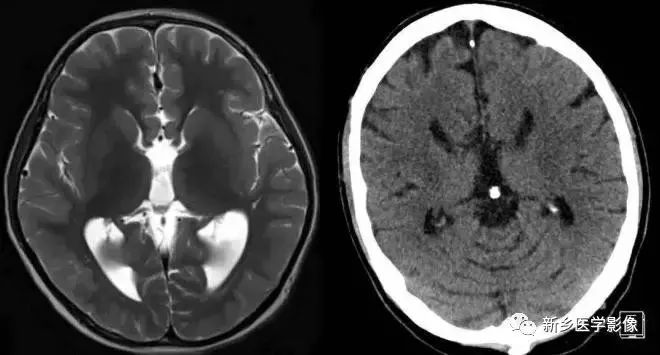
四、三脑室及侧脑室同时扩大
1.导水管狭窄:
中脑导水管是脑室系统最狭窄的通道,也是脑脊液循环受阻最常见的部位。
导水管狭窄的病因比较复杂、主要包括:①先天发育性狭窄②导水管周围胶样变③导水管粘连等。先天发育性狭窄可呈线样狭窄、分叉样狭窄或横膈膜形成。导水管周围胶样变多为炎症所致,主要见于宫内先天感染后,尤其是弓形体感染。
导水管粘连主要见一于颅内感染和出血后,可于胚胎期发生,也可见于出生后任何年龄。导水管粘连所致狭窄多位于导水管远端。狭窄段长度通常为2一5mm,狭窄近端异水管可呈喇叭口样扩张。
导水管狭窄时,三脑室扩大常很显著,三脑室前部视隐窝和漏斗隐窝扩张或消失,三脑室后部松果体隐窝和松果体上隐窝明显后突,向小脑上池疝入。严重者可压迫小脑。
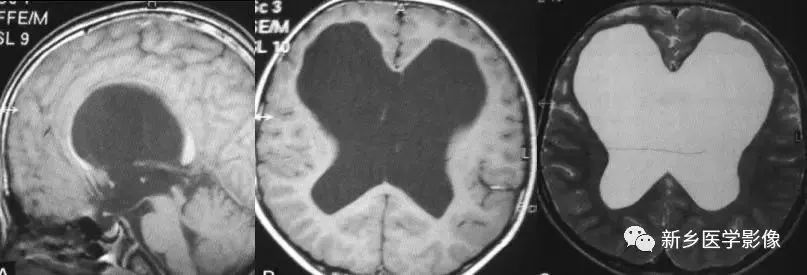
2.小脑扁桃体下疝畸形
又称Chiari’s畸形,即小脑扁桃体下移到椎管内,延髓、四脑室延长并部分向下移位。可分为3型。各型均常有脑积水表现。三脑室及侧脑室扩大。Ⅰ型:若仅有小脑扁桃体下移。扁桃体下缘低于枕大孔连线5mm以上,无脑干及四脑室改变者为Ⅰ型。Ⅱ型:除小脑扁桃体下移外,同时有四脑室部分或全部降入枕大孔以下者为Ⅱ型。Ⅲ型:全小脑及四脑疝入枕大孔以下者为Ⅲ型。
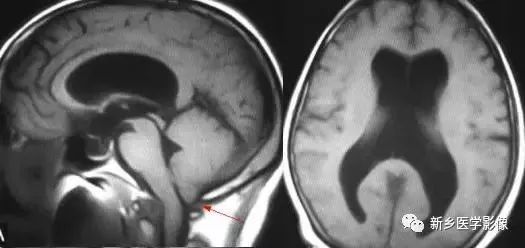
五、全部脑室扩大
1.交通性脑积水
又称脑室外梗阻性脑积水,是由四脑室出口以后脑脊液循环通路障碍所致的脑积水。常位于蛛网膜下腔,以基底池最常见。主要原因包括脑膜炎、蛛网膜下腔出血、脑膜转移、外伤、静脉窦血栓、颅脑手术后和脑脊液吸收功能障碍等。临床表现主要由颅压增高所引起. 可表现有头痛、呕吐、复视和视乳头水肿等。交通性脑积水时.第四脑室扩大通常出现较晚,故早期时,可仅表现有侧脑室和三脑室扩大。主要应与导水管狭窄之梗阻性脑积水区别。MR矢状位T1加权图直接观察导水管有无狭窄的最好方法。另外需要与普遍性脑萎缩区别,脑萎缩时沟脑裂增宽,而脑积水时脑沟变窄消失或正常。另外.三脑室扩大不明显脑室扩大比较明显。脑萎缩时三脑室扩大较明显。到晚期,交通性脑积水出现整个脑室系统普遍扩大而脑沟正常或变窄消失。
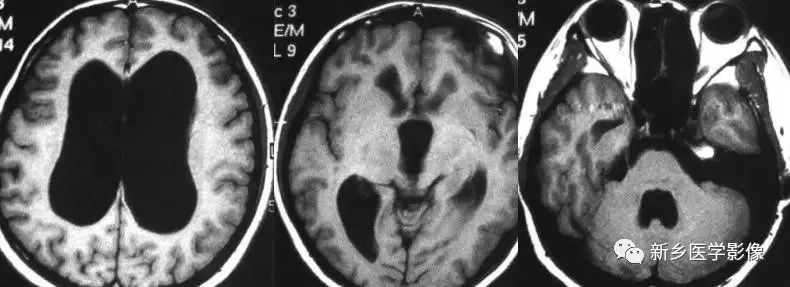
2.正常压力性脑积水
实际上是交通性脑积水的一种特殊类型.又称隐匿性脑积水、低位性脑积水或慢性交通性脑积水。脑脊液压力正常的原因可能是由①脑脊液代偿性分泌减少,而吸收加强,使脑脊液压力处于相对正常状态;②脑脊液经室管膜吸收。使脑脊液压力保持止常;③脑实质的张力降低。正常压力性脑积水可能由隐匿性蛛网膜下腔出血或感染所致.也可能继发于其他颅脑疾病。包括蛛网膜下腔出血、脑外伤、炎症、颅脑手术后粘连等。正常压力性脑积水常见于50岁以后。临床表现实际上是慢性交通性脑积水的表现加上脑萎缩的表现,其典型临床表现为痴呆、步态异常和二便失禁,并呈进行性加重。少数病例还有肢体和短暂意识障碍。CT和MR检查表现为普遍性脑室扩大,但脑沟并不变窄或消失。或表现有脑沟脑裂增宽,与脑萎缩鉴别很困难。正常压力性脑积水也可仅表现有脑萎缩,而没有脑室扩大,所以,临床症状典型时,仅有脑萎缩表现并不能除外正常压力性脑积水的诊断
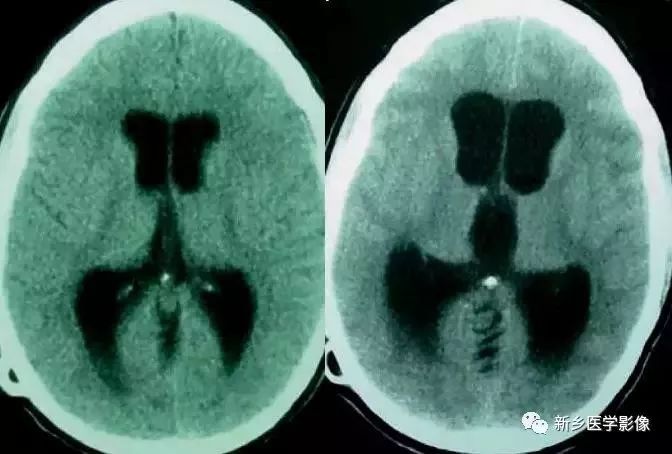
3.四脑室出口阻塞
单纯四脑室出口阻塞多是由粘连引起。临床少见。粘连后引起梗阻性脑积水,全部脑室均扩大,但通常以四脑室扩大最为显著,平扫常难以与四脑室内囊肿区别。最有效的鉴别的办法是行脑室照应CT检查,若为囊肿时表现为四脑室内圆形充盈缺损,四脑室出口粘连阻塞时,四脑室内充满脑脊液
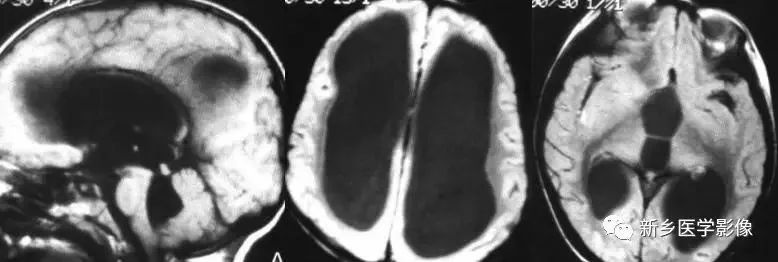
4.Dandy-Walker’s综合症
又称第四脑室中侧孔先天性闭锁。在胚胎早期期,第四脑室正中孔及侧孔闭寒,导致四脑室呈囊性扩张,并伴有小脑蚓部及半球发育不良,扩张之四脑室向后发展,并与枕大池相连,使后颅窝扩大,小脑幕抬高。本病出现脑积水通常见于婴儿期,或者出生后即存在,但到成人期才发病。
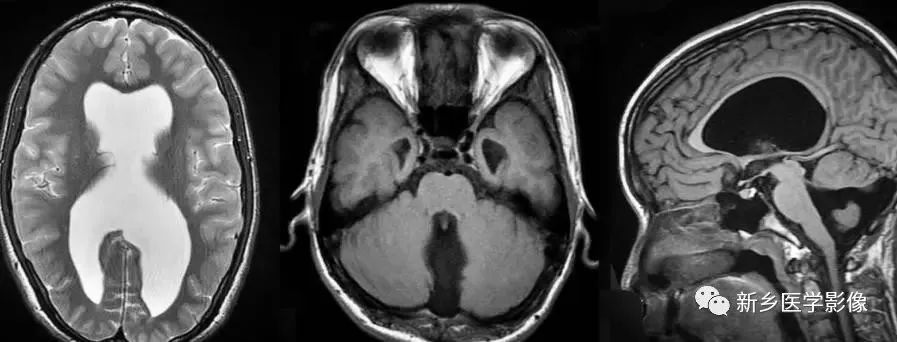
5.四脑室内囊肿
四脑室囊肿可引起整个脑室系统梗阻扩大。
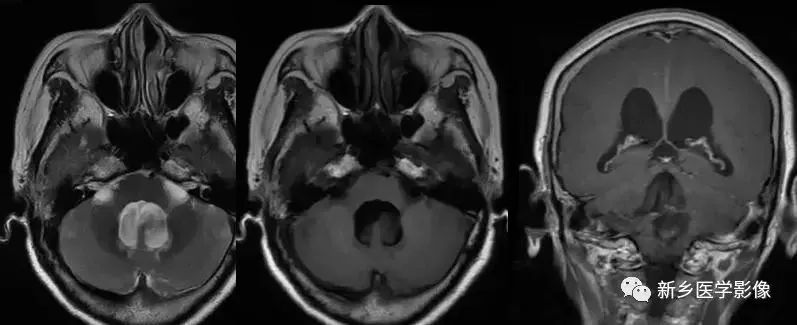
6.孤立性第四脑室
当导水管和第四脑室出口严重狭窄或完全阻.塞时,四脑室则被孤立起来,这种情况称为孤立性第四脑室。


