Nature Communications:AD相关PKCα突变加速认知损伤
时间:2022-12-15 18:00:24 热度:37.1℃ 作者:网络
阿尔茨海默病(AD)是老年人中最常见的神经退行性疾病,其特点是突触退化、神经元死亡,并最终导致参与学习和记忆的大脑区域面积缩小。AD的大脑因存在导致认知损害的神经纤维缠结和细胞外淀粉样β(Aβ)斑块而与其他神经退行性疾病区分开来。然而,AD的分子机制仍然难以捉摸。
近期,《Nature Communications》期刊上发表了题为“Enhanced activity of Alzheimer disease-associated variant of protein kinase Cα drives cognitive decline in a mouse model”的论文,作者团队利用基因编辑将M489V功能获得型变体引入内源性PKCα,以确定这种单一的氨基酸变化,适度提高的激酶活性,是否足以驱动小鼠模型中的AD病理。
生物化学、磷酸化蛋白质组学、电生理学和行为学研究显示,这种AD变体的存在所导致的PKC活性的提高,使大脑中PKC底物的磷酸化增加,神经元变性,增强Aβ驱动的突触抑制以及认知能力的下降,这在AD的转基因小鼠模型中表现得更为明显和迅速。这些数据表明PKCα活性的增强足以驱动小鼠模型的认知能力下降,并支撑将PKCα的抑制作为AD的一种潜在治疗方法。
 研究背景
研究背景
目前,AD发生发展最具说服力的假说之一是β和γ分泌酶对淀粉样前体蛋白(APP)的不当处理所产生的Aβ 42肽的异常积累。通常,可溶性Aβ低聚物被认为是引发疾病及其伴随症状的神经毒性物质。然而,Aβ斑块是相对惰性的,但它可以作为可扩散的低聚物的储存库。APP和presenilin基因(PSEN1和PSEN2)是第一个显示出有与AD相关变体基因,并对进展性淀粉样蛋白级联假说和建立Aβ斑块的错误处理和沉积与AD发展之间的联系至关重要。
尽管这些变体在AD病例中占的比例相对较低,但它们的鉴定为探索与该疾病相关的复杂遗传学找到方法。APOE的多态性在早期也被确定为是AD的主要危险因素。全基因组测序工作正在确定与AD风险相关的其他遗传变异,包括TREM2、PLCG2和ABI3等基因的多态性。在最近一次寻找与AD相关的罕见功能变异中,对来自NIMH队列的410个家庭中受影响和未受影响的兄弟姐妹的全基因组测序数据进行分析,发现了PKCα的变异。其中一个变体M489V(rs34406842,在gnomad中的小等位基因频率为0.00095)只存在于4个家庭的受影响成员中,在未受影响的成员中不存在,并与AD患者的情感状态有关联。蛋白激酶的高可成药性使PKCα成为AD潜在的治疗靶点。
PKCα被称为传统PKC,属于依赖Ca2+和二酰甘油(DG)的PKC同工酶。这些Ser/Thr激酶从受体介导的膜磷脂水解中转导信号,从而产生它们的激活剂,即Ca2+和DG。传统的PKC同工酶在维持细胞平衡方面起着关键作用,它们的活动经过微调,调节着细胞死亡和生存之间的平衡。它们通过一系列有序的磷酸化形成一种稳定的、自抑制的构象,从而对第二信使作出快速和可逆的反应。异常的PKC如果没有适当的自我抑制,就会通过质控途径降解。尽管PKC历来被认为是致癌的,但最近的分析表明,PKC同工酶具有抑制肿瘤的作用。值得注意的是,与癌症有关的突变通常是功能缺失表型,而PKC同工酶蛋白水平的提高使许多癌症患者的生存率提高。因此在癌症中抑制PKC是不成功的,在某些情况下会使病人的结局恶化。识别出与AD共存的PKCα活性增强变体,就有可能使AD患者从原本用于癌症临床试验的PKC抑制剂中获益。
与癌症明显不同的是,PKCα的功能获得型突变已被证明与AD共分离。在LOAD的家庭中PKCα的高渗透性种系变异,表明PKC的功能失调可能是AD的致病因素。与PKC在AD病理中的作用一致,磷酸化蛋白质组学研究已经确定PKC底物包括MARCKS磷酸化增强,是AD发展的主要事件之一。此外,电生理学研究已经确定,PKCα是Aβ依赖性突触抑制的必要条件,其机制需要PKC同工酶的PDZ配体。这种PDZ配体将PKCα锁定在PSD95、SAP97和PICK1支架上,后者也是Aβ依赖性突触抑制的必要条件。迄今为止,所有与AD相关的PKCα变体都是功能获得型。对存在于四个不相关家族中的一个变体M489V PKCα的生化分析显示,激活环中Met到Val的替换使该酶的内在催化活性增加了30%,而不影响自抑性构象的稳定性,因此,逃避了细胞对异常活性PKCα的正常降解。综上所述,这些结果支持一个模型,即PKCα在突触后支架上的活性介导了Aβ对AD的影响,活性增强加剧了AD的病理。确定与AD相关的PKCα的突变是否足以驱动AD的病理变化,将有助于了解PKCα的抑制是否是AD的潜在治疗策略。
结 果
结果一:M489V PKCα小鼠大脑磷酸化蛋白组的改变
PKCα M489V是一种与AD相关的突变,已被证明比野生型(WT)PKCα的催化活性更高,其机制是不损害其稳定性,使其能够逃避细胞对异常活性PKCα的稳态调控。为了了解由于PKCα活性增加而在大脑中发生的磷酸化事件,我们分离了携带WT和M489V PKCα的C57BL/6小鼠3月龄时的大脑,并对其进行了磷酸化蛋白质组学分析。简而言之,我们从脑裂解液中提取和消化蛋白质,用TiO2富集磷酸肽。用TMT 10-plex试剂标记未富集的肽和磷酸肽,并通过液相色谱-MS2/MS3(LC-MS3)进行分析,对蛋白质组和磷酸化蛋白组进行量化(图1a)。
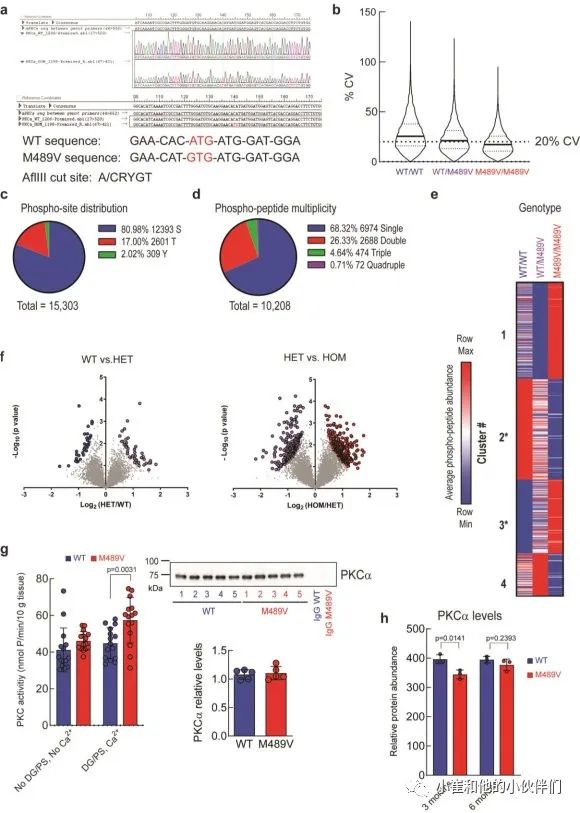
我们对每个样品的10208个磷酸肽进行量化,代表1899个蛋白质,肽和蛋白质水平的错误发现率(FDR)小于1%(数据集S1)。样品内变异系数的中位数平均约为20%,表明变异最小(补充图1b)。磷酸位点的分布是80.98%的磷酸丝氨酸(pS),17.00%的磷酸苏氨酸(pT)和2.02%的磷酸酪氨酸(pY),大多数肽在单位点(68.32%)和双位点(26.33%)上磷酸化,较少在三位点(4.64%)和四位点(0.71%)上磷酸化(补充图1c, d)。
M489V PKCα变体的存在诱发了各种磷酸化蛋白的明显变化(图1b和补充图1f)。具体地说,M489V PKCα诱发了总共829个磷酸肽的显著变化。其中,270个独特蛋白质的430个肽的磷酸化增加(红色),261个独特蛋白质的399个肽的磷酸化减少(蓝色)(图1b;补充表1列出了PKCα M489V大脑中磷酸化增加的前25个肽)。在携带M489V PKCα变体的小鼠大脑磷酸化增加的肽中,我们发现一种PKC底物MARCKS的Ser156和Ser163的磷酸化增加(图1b,c)。特别的是,MARCKS的磷酸化增加,与先前报道的纯化的PKCα M489V与WT相比催化活性增加是一致的,从M489V小鼠的脑裂解液中免疫沉淀的PKCα也观察到这种增加(补充图1g)。重要的是,对用于质谱分析的小鼠同胞的脑裂解物的免疫印迹分析先前已经确定,PKCα的数量不受WT和M489V小鼠的影响(也见补充图1h),这与作者团队的生化研究一致,该研究表明PKCα的M489V突变不改变该蛋白的稳态水平。
接下来,作者用k-means聚类法评估了PKCα M489V在杂合子或同合子中引起的磷酸化蛋白组的变化,其磷酸化程度在三组中发生了明显的变化(补充图1e)。PKCα M489V诱导的磷酸化蛋白组分为四个不同的簇,其中第2簇(C2)和第3簇(C3)分别显示了基因剂量依赖的磷酸化减少和增加的肽(图1d,e,补充图1e)。为了探索与PKCα中的AD变体所产生的增强的激酶活性有关的生物学功能,作者进行了gene ontology(GO)富集分析,以比较C2(底物的磷酸化以基因剂量依赖方式减少)和C3(磷酸化以基因剂量依赖方式增加)中所代表的细胞区系(图1d,e)。突触后密度、突触、细胞连接和突触后膜显著地富集在多肽组中,这些多肽的磷酸化以基因剂量依赖的方式(C2)下降(图1d,右)。然而,其磷酸化随着变体(C3)的功能而增加的肽在细胞骨架、细胞质和微管功能中显示出明显的富集(图1e,右)。这些数据与PKCα的功能增强以增加直接底物(如MARCKS)的磷酸化以调节细胞骨架功能,并通过增强磷酸酶活性或抑制针对这些底物的激酶,间接减少作为突触关键调节者的底物的磷酸化是一致的。
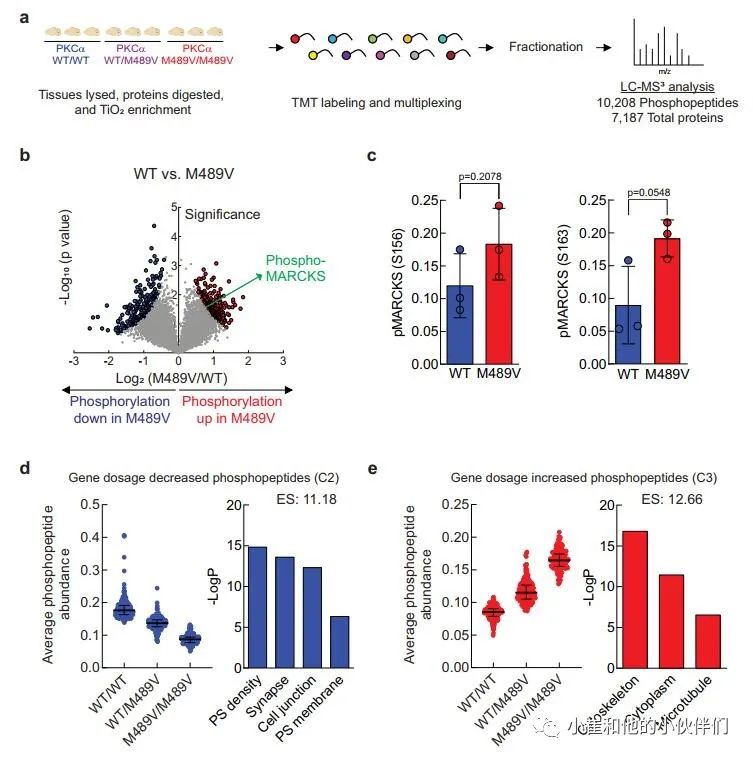 图1 3个月大的WT小鼠和C57BL/6中携带PKCα M489V
图1 3个月大的WT小鼠和C57BL/6中携带PKCα M489V
突变的小鼠大脑的磷酸蛋白质组学分析
 补充图1 野生型小鼠和C57BL/6背景下携带PKCαM489V
补充图1 野生型小鼠和C57BL/6背景下携带PKCαM489V
突变的小鼠大脑中的磷酸蛋白质组学分析
结果二:M489V PKCα小鼠海马神经元的棘突密度下降
鉴于突触后蛋白是M489V大脑磷酸化蛋白组分析中变化最大的蛋白之一,作者研究了M489V小鼠的突触与WT小鼠相比是否有形态上的改变。大脑中兴奋性突触的突触后部分是由树突棘组成的。棘突的形态是提示相关突触稳定性,可塑性和强度的一个指标。大脑中的大部分兴奋性突触存在于树突棘上,因此,海马区树突棘密度的调节被认为在学习和记忆中起着核心作用。为了更好地了解PKC活性增强与神经变性之间的联系,作者检测了4.5月龄的雄性WT小鼠和PKCα M489V变体小鼠的海马神经元的棘突密度。通过对神经元突起的荧光染色,作者观察到与WT小鼠相比,M489V同型小鼠的棘突密度略有下降(9.81±0.02%),但具有统计学意义(图2a, b)。
此外,对分离的海马进行的蛋白印迹分析显示,与WT相比,M489V的样品的PKCα中,PKC底物的磷酸化普遍增加(图2c)。MARCKS在Ser159/Ser163的磷酸化(图2c)也在PKCα M489V海马样本中增加,这与全脑磷酸化蛋白组学数据(图1b,c)相一致,并且以前也通过Western blot验证过。此外,与WT小鼠相比,PKCα M489V海马中的细胞外信号调节激酶(ERK)1/2(一种MAP激酶家族成员)的磷酸化增强(图2c)。总之,PKCα M489V变体小鼠显示出棘突密度降低,以及调节神经元的蛋白质的磷酸化增强,这是阿尔茨海默病的一个重要标志。
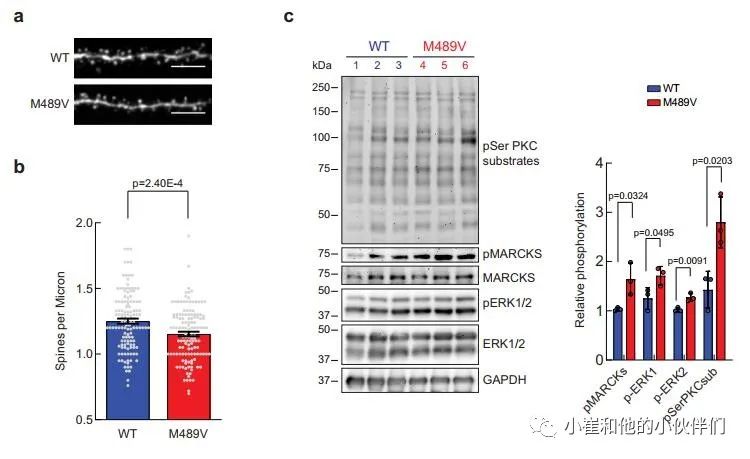 图2 AD相关PKCα M489V突变降低脊柱密度,增加海马PKC底物磷酸化
图2 AD相关PKCα M489V突变降低脊柱密度,增加海马PKC底物磷酸化
结果三:C57BL/6中的PKCα M489V小鼠的认知能力受损
由于海马的棘突密度下降与学习能力下降相关,作者接下来评估PKCα M489V变体是否会影响认知能力。作者检测了WT和M489V小鼠在Barnes迷宫中的行为表现,这个测试被广泛用于评估AD的空间学习和记忆。在这个测试中,小鼠被训练使用迷宫周围的不同线索,在一个圆形平台周边的20个孔中找到一个逃生盒(图3a)。记录小鼠在每个象限(目标象限与非目标象限)寻找洞的时间。相对于迷宫的其他区域,认知完整的小鼠在目标象限花费更多时间,而认知受损的小鼠不能很好地区分四个象限。所有年龄段的WT小鼠在这个测试中表现良好,在目标象限的时间大约是其他象限的两倍(图3b-d)。与此形成鲜明对比的是,随着年龄的增长,携带M489V突变的小鼠在目标象限的时间逐渐减少,以至于在12个月时,它们不再能区分目标象限和其他象限(图3d)。甚至在3个月大时,它们识别目标象限的能力也比WT小鼠低(在目标象限的时间分别为34±2%和41±5%)。在目标象限的时间减少并不是运动能力受损的反映,因为在Barnes迷宫探针测试和独立的活动测试中,WT和M489V小鼠的活动水平是一样的(补充图2a和补充图2b)。此外,M489V小鼠并没有在明/暗测试评估中表现出焦虑的增加(补充图2c)。因此,与WT小鼠相比,M489V小鼠的学习和记忆受损,但在活动能力或焦虑方面没有改变。这些结果表明,与AD相关的PKCα突变M489V足以导致C57BL/6小鼠的认知能力下降。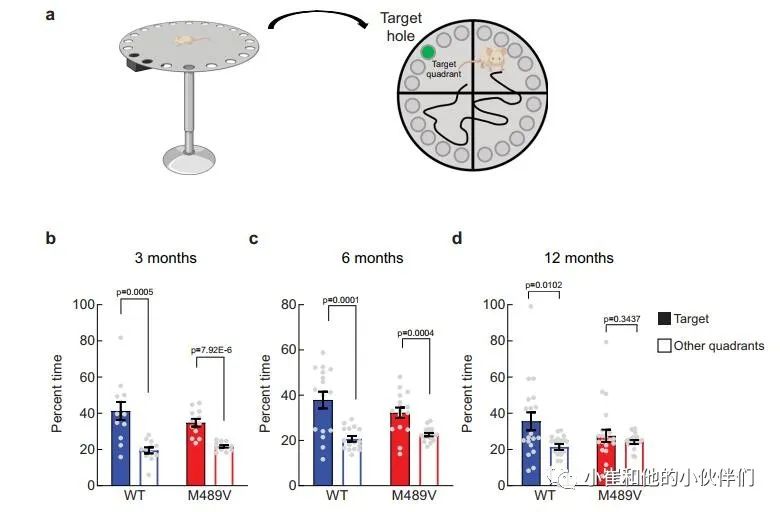 图3 Barnes迷宫试验中C57BL/6 PKCα M489V/M489V
图3 Barnes迷宫试验中C57BL/6 PKCα M489V/M489V
小鼠的空间学习和记忆受损
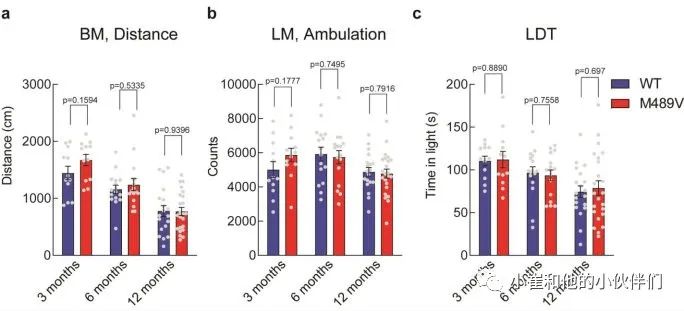 补充图2 WT和M489V C57BL/6小鼠的其他行为测试
补充图2 WT和M489V C57BL/6小鼠的其他行为测试
结果四:PKCα M489V小鼠的神经元中Aβ诱导的突触抑制增强
阿尔茨海默病的特点是存在神经元斑块,它主要由APP蛋白裂解产生的Aβ肽组成。众所周知,Aβ聚集物会使突触退化并损害记忆形成。由于PKCα是Aβ诱导突触抑制所必需的,作者推断,PKCα M489V AD突变的活性增强可能会增强对Aβ的电生理反应。用表达CT100(APP被β分泌酶裂解的产物)的Sindbis病毒载体感染WT或M489V小鼠的海马切片。由于CT100的表达,在被感染的神经元中,由于γ-分泌酶的处理使Aβ肽的产量增加,导致突触抑制。
感染后18-24小时,通过从两个相邻的锥体海马CA1神经元(一个被感染,一个未被感染)获得全细胞记录来研究突触传递;用电刺激Shaffer侧枝轴突来激发AMPAR介导的兴奋性突触后电流(EPSCs)(图4a)。这种细胞对的记录使得人们能直接比较Aβ升高对突触传递的影响,因为无论刺激强度如何,针对受感染和未受感染细胞的激活Shaffer侧枝轴突的数量平均是一样的。正如预期的那样,在WT和M489V切片中,与未感染的邻近神经元相比,表达CT100的神经元显示出突触抑制。
然而,这种影响在M489V小鼠的切片中更为明显(WT小鼠有26±12%的抑制;M489V小鼠有55±6%的抑制;P<0 . 0 5;图4b-d)。这些结果表明,神经元中Aβ依赖性的突触抑制因更活跃的PKCα的存在而增强,PKC活性增加驱动与AD相关的突触下降。
 图4 AD相关的PKCαM489V突变加剧了Aβ诱导的突触抑制
图4 AD相关的PKCαM489V突变加剧了Aβ诱导的突触抑制
结果五:M489V PKCα变体的tg-AD小鼠的磷酸化蛋白组学分析
与WT神经元相比,PKCα M489V海马神经元由Aβ诱导的突触抑制增强,功能获得型的PKCα突变是否会加剧APP的存在所造成的影响。为此,作者将M489V突变引入到带有瑞典突变的APP转基因的B6;SJL小鼠上(tg-AD);这种成熟的小鼠模型具有AD的易感性,这是由于β分泌酶对突变体APP的异常处理造成Aβ水平升高的结果。作者首先研究了将PKCα M489V突变引入带有APPswe转基因的tg-AD小鼠模型是否会影响大脑中可溶性和不可溶性Aβ的存在。大脑被均质化,提取不同的Aβ并通过酶联免疫吸附试验(ELISA)进行分析。
Aβ-40和Aβ-4 2的水平,以及Aβ-42/Aβ-40的比例,在WT PKCα或M489V变体的小鼠中是相同的(补充图3a-c)。这些结果被组织化学分析所证实,WT tg-AD和M489V tg-AD小鼠的大脑都呈现出类似的Aβ斑块水平(补充图3d)。这表明,PKCα活性增强导致神经变性和认知能力下降的分子事件是在Aβ产物的下游或非Aβ依赖的。
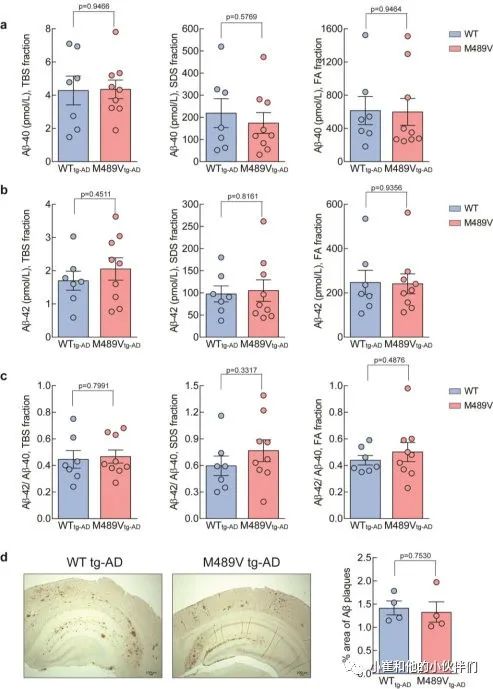
补充图3 WT小鼠和携带AD转基因小鼠PKCα M489V
突变的小鼠大脑中的淀粉样β蛋白水平
鉴于PKCα M489V变体并不影响tg-AD小鼠模型的Aβ产生,作者推断它是Aβ下游的信号传导,并可能在APPswe转基因诱导的变化基础上进一步降低磷酸化蛋白组。因此,作者研究了PKCα M489V是否以类似于在非tg-AD背景下观察到的改变的方式改变了AD小鼠模型的磷蛋白组。在4.5和6个月大时,从携带WT或M489V PKCα的小鼠中存在APPswe转基因(WT tg-AD/M489V tg-AD)或不存在(WT非tg-AD/M489V非tg-AD)的大脑组织被分离出来。组织的处理和分析如图1a所示,以量化蛋白质组和磷酸化蛋白组(图5a)。
作者在每个样品中量化了约12000个磷酸肽,代表约6500个蛋白质,肽和蛋白质水平的FDR小于1%(数据集S2)。样品内变异系数的中位数平均<20%,表明变异最小(补充图4a)。与以前的实验一致,磷酸化位点的分布是77.41%的磷酸丝氨酸(pS),19.40%的磷酸苏氨酸(pT)和3.19%的磷酸酪氨酸(pY)(补充图4b),大多数肽的磷酸化位点是单位(73.60%)和双位(23.10%),较少的是三位(2.96%)和四位(0.35%)(补充图4c)。
作者接下来分析了不同群组小鼠的APP和PKCα水平。正如预期的那样,所有tg-AD小鼠(WT tg-AD/M489V tg-AD)的APP数量持续上升。值得注意的是,每组PKCα的相对丰度都是一样的(图5b)。这些数据突显了这种分析的可重复性,并验证了作者的生化研究,表明M489V突变并不改变体内PKCα的稳定性/稳态水平。全脑裂解液的Western blot分析显示,APP转基因增加了PKC的活性,与对照组小鼠相比,携带APP转基因的小鼠大脑中PKC底物的磷酸化程度增加(图5c)。这与磷酸化蛋白组学中的分析显示了大量底物的增加一致(在tg-AD小鼠中PKC-M489V增强的前25个磷酸基点见补充表2和3)。
在AD小鼠模型中引入PKCα M489V突变导致的磷酸化蛋白组的变化,使用k-means聚类对28,084个磷肽进行分析,发现有9个不同的簇(补充图4d)。与年龄有关的磷酸化蛋白组簇为三个不同的组。C1、C6和C8。C1和C8包含的肽的磷酸化随着年龄的增长而增加,而C6包含的肽的磷酸化随着年龄的增长而适度减少。M489V仅在早期年龄组(4.5个月)的APP大脑磷蛋白组的C2和C4群中引起了磷酸化的增加。C3和C5群包含的多肽在4.5个月时磷酸化程度下降。C9是特别令人感兴趣的(图4d),因为它所包含的蛋白质的磷酸化在4.5个月到6个月大的WT PKCα小鼠中只略有增加,但在携带PKCα M489V变体的小鼠中则随着年龄的增长而大幅增加(图5d,左)。
作者推断,这组蛋白质包括在tg-AD背景下,其磷酸化随着年龄的增长而增加,而PKCα的突变则加剧了这种情况。为了探索与这一独特的磷蛋白组相关的生物学功能,作者用GO分析法分析了在C9中被过度代表的生物学过程。学习、轴突发生和突触囊泡内吞作用在C9中明显富集,蛋白质磷酸化和树突发育也是如此(图5d,右)。此外,串联分析显示,这些蛋白质中有许多是mTOR信号通路、丝裂原激活蛋白激酶(MAPK)信号通路的一部分,并参与了神经元的投射(图5e);在WT背景下,神经元投射过程也被M489V变体明显扰乱(例如,图1d,e)。研究ERK1/2的磷酸化以进一步确定PKCα突变对这些途径的影响。以前曾描述过,APP的表达和暴露于低聚物Aβ肽会增强Ras/ERK信号,增加异常增殖和随后的神经退行性。免疫印迹分析显示,与WT非tg-AD小鼠相比,M489V非tg-AD小鼠和tg-AD小鼠的脑裂解液中ERK1/2的磷酸化都有所增加(图5f)。这种增加也被磷蛋白组学分析所捕获(图5g)。因此,PKCα M489V突变或APP转基因都会增强Erk的信号传导。总的来说,磷蛋白组学分析表明,在4.5和6个月大的携带APP转基因的小鼠大脑中,PKCα催化活性的轻微增强会诱发蛋白质磷酸化的变化。这种信号的改变可能导致细胞周期失调、异常的增殖信号以及随后的老年记忆和学习障碍。
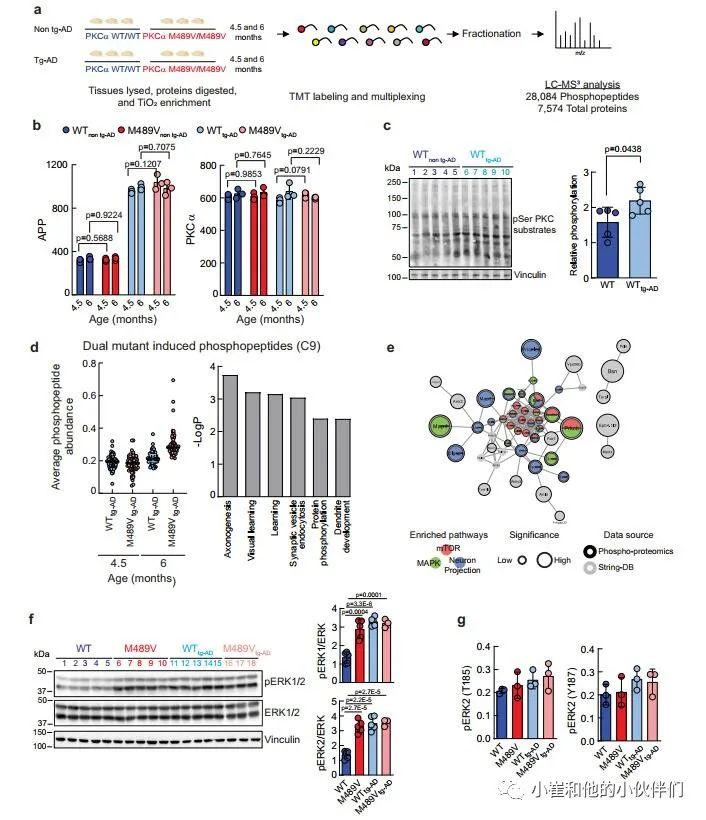
图5 来自WT小鼠和B6;SJL上带有APP转基因携带瑞典突变(APPswe)的PKCαM489V突变(红色)小鼠的脑磷蛋白质组学分析
结果六:PKCα M489V加速了tg-AD小鼠的认知障碍
上述研究表明,PKCα M489V AD变体的存在足以影响C57BL/6小鼠3个月时的大脑磷酸化蛋白组和12月龄时的学习能力。为了阐明APP的存在是否会加剧或加速这种对认知的影响,作者使用了空间学习和记忆的评估,在4.5(图6a)、6(图6b)和12月龄(图6c)时,携带PKCα M489V突变(红色)和WT对照(蓝色)的没有(WT和M489V)或有(W T tg-AD / M489V tg-AD)的APPswe转基因的小鼠进行Barnes迷宫试验。将这种PKCα突变引入没有APP转基因的B6;SJL(非tg-AD),在测试的早期年龄都没有引起认知障碍(图6a,b),但在12个月大时有明显缺陷(图6c)。
因此,正如C57BL/6小鼠所报告的那样,仅PKCα突变就足以导致非tg-AD小鼠的认知障碍(图3d)。在WT tg-AD/M489V tg-AD小鼠中,APPswe转基因的存在并没有导致4.5个月大的小鼠Barnes迷宫表现的缺陷(图6a),但它确实在12个月时产生了认知缺陷(图6c)。令人震惊的是,在6个月时,WT PKCα的tgAD小鼠(WT tg-AD)具有正常的认知能力,但M489V突变(M489V tg-AD)的存在消除了小鼠对目标象限(填充条)和其他象限(开放条)的辨别能力(图6b)。因此,PKCα M489V突变加速了AD小鼠模型的认知能力下降。需要强调的是,M489V或M489V tg-AD小鼠在Barnes迷宫探测试验或运动活动试验中都没有表现出活动水平的降低,也没有在明/暗试验中增加焦虑样行为(补充图5)。
因此,这种与AD相关的PKCα的突变,使该酶的催化活性提高了30%,当与APPswe转基因配对时,6个月大的小鼠认知能力急剧受损(图6b),在12个月大时足以单独影响学习和记忆(图3d和图6c)。这些发现表明:1)PKCα的M489V突变足以单独导致小鼠的行为缺陷;2)该小鼠品系中与APPswe相关的认知能力下降在同时携带PKCα突变的小鼠中会加速。
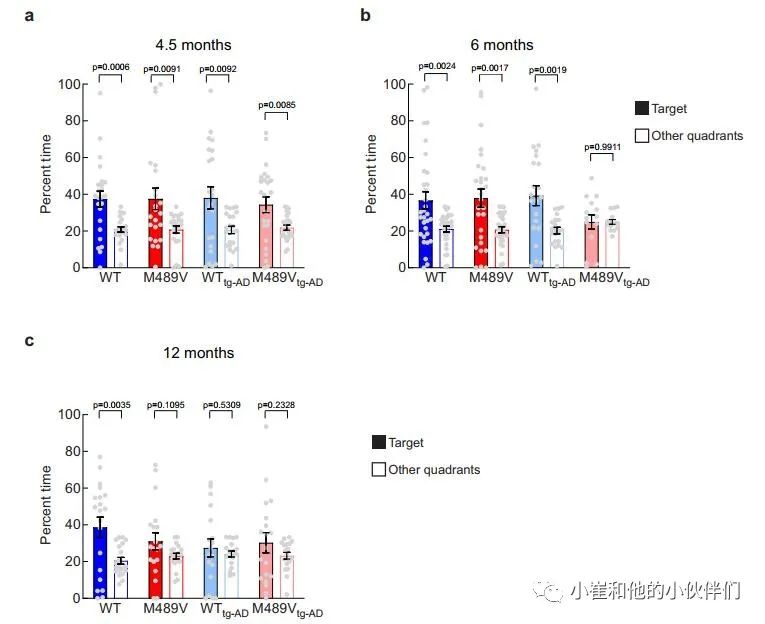 图6 PKCαM489V小鼠+AD转基因小鼠Barnes迷宫试验中的空间学习受损
图6 PKCαM489V小鼠+AD转基因小鼠Barnes迷宫试验中的空间学习受损
结果七:人阿尔茨海默病患者大脑中的PKCα水平增加
上述数据表明,由罕见但高度渗透的PKCα变体导致的PKCα信号增强足以损害认知能力。鉴于PKC信号的升高已被检测为AD病理学中最早的事件之一,作者想知道PKCα的稳态水平是否会在AD中升高,从而导致信号输出增强。在这方面,细胞中PKC蛋白的数量调节其信号输出,较高的稳态水平导致较高的信号输出;例如,相关的同工酶PKCβII已被证明在抑制致癌信号方面是不足的。
因此,作者用免疫印迹法评估了已故AD患者或对照者大脑额叶皮层中PKCα蛋白的水平,以衡量PKCα信号输出的增强是否与AD有关。该分析显示,在AD患者死后的大脑中,PKCα的稳态水平有提升20%并具有统计学意义(图7a)。此外,作为PKC激活的另一个标志,我们评估了SAP97上T656的磷酸化状态,这是一个先前确定的PKCα磷酸化位点。与对照组患者相比,AD患者大脑中T656的磷酸化状态增加了约4倍,这与PKC的高活性相一致(图7b)。这些数据显示,PKCα在人类AD大脑中普遍上调,导致底物磷酸化增强。
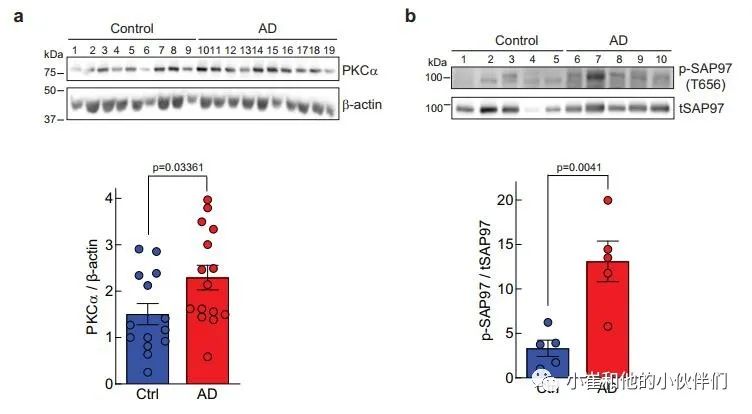 图7 阿尔茨海默病患者额叶皮层PKCα水平和SAP97磷酸化水平升高
图7 阿尔茨海默病患者额叶皮层PKCα水平和SAP97磷酸化水平升高
小 结
对蛋白激酶C(PKC)同工酶的活动进行精确的调整对维持细胞的平衡至关重要,其功能缺失型突变通常与癌症相关,而同功酶PKCα的功能获得型突变则与阿尔茨海默病(AD)相关。作者发现突变体PKCα M489V的活性增强,足以使小鼠模型重构大脑磷酸化蛋白质组,驱动突触变性并损害认知能力。该突变导致催化活性大约增加30%,而不改变开/关激活动态或稳定性,催化活性的增强足以导致所观察到的生化、细胞结果和最终认知影响。对PKCα M489V小鼠海马神经元的分析显示,与野生型小鼠相比,淀粉样β诱导的突触抑制增强,脊柱密度降低。行为学研究显示,这种突变本身就足以导致认知能力下降,再加上小鼠的AD模型,会进一步加速认知能力的下降。蛋白激酶的可成药性使PKCα成为AD潜在的治疗靶点。

