脑损伤患者的脑氧输送和消耗
时间:2023-04-03 16:03:34 热度:37.1℃ 作者:网络
简介
生物体的生存取决于氧气的输送和利用,以维持能量和有毒氧化剂产生的平衡 。这种调节对中枢神经系统 (CNS) 至关重要。脑组织呈现出一种特殊的动态能量消耗。能量类似物最高效的代谢过程是氧化磷酸化,与耗氧量有关 。早在 1890 年,罗伊 (Roy) 和谢林顿 (Sherrington) 就观察到,增加的神经元活动会增加能量消耗以及代偿性代谢和脉管系统反应,进而改善神经元的功能 。因此,脑组织中的氧含量是影响神经和神经胶质细胞功能的关键因素。
脑损伤是全世界发病率和死亡率的常见原因,尤其是在年轻人中 。继发性脑损伤发生在事件发生后的数小时、数天或数周内,并且与致命结果相关 。继发性损伤可能由脑代谢受损、缺血、颅内高压和氧浓度紊乱(如缺氧)介导。缺氧和低血压的结合与极高的死亡率有关 。最近的数据强调了临床方案在改善氧输送和降低创伤性和非创伤性脑损伤患者死亡率方面的重要作用. 临床方案根据生理过程指导氧补充规则,例如增加氧供应(通过监测平均动脉压 (MAP) 和颅内压 (ICP)、脑血管反应性和氧容量)和减少氧需求(通过药物镇静和昏迷或亚低温)。因此,监测脑组织氧 (PbtO 2 ) 等氧浓度是脑损伤临床实践的一个重要方面 。此外,监测局部和全身组织的平均动脉压和氧合对于氧合和最终结果至关重要 。
这篇综述的目的是讨论不同条件下大脑中的氧代谢。
氧输送和自动调节
大脑的重量仅占人体的 2%,但脑组织使用 25% 的葡萄糖和约 20% 的氧输送来正常运作 。耗氧量为3.5毫升氧气/100克组织/分钟;因此,调节血流和向脑组织输送氧气对于脑功能至关重要 。重要的是,神经元消耗的能量的 75-80% 用于突触以恢复在去极化过程中丢失的神经元膜电位 . 人体通过动脉血向大脑持续供应氧气,并通过扩散输送到脑组织。扩散与脑组织的氧传导率有关,由毛细血管的几何形状(距离和面积)和组织的新陈代谢(从毛细血管到组织的氧梯度)决定 。 氧摄取取与血流量成反比(当新陈代谢恒定时),与新陈代谢(当流量恒定时)和组织与毛细血管之间的面积成正比。因此,氧输送的减少增加了氧摄取。应该注意的是,当脑血流量 (CBF) 减少 50–60% 时,随之而来的氧摄取升高不足以维持适当的脑氧合和恒定的脑氧代谢率 (CMRO2) 。 因此,脑氧输送由血氧含量和脑血流量决定。在生理条件下,由于大动脉对血管阻力的贡献,以及实质小动脉对相当大的基础张力的影响,大脑中的总血流量是恒定的。
脑血流量的自动调节是使大脑能够通过改变灌注压来维持相对恒定的血流量的机制。在血压正常的生理状态下,随后的脑灌注压 (CPP) 在 60 至 160 mmHg 的范围内,CBF 维持在每 100 g 脑组织每分钟 50 mL。超出此范围后,自动调节功能将丧失,CBF 开始以线性模式依赖于 MAP。CPP 低于下限 50 mmHg 会导致脑缺血 。CBF 的这种减少通过从血液中摄取取更高的氧来补偿。
通过针对最佳、接近大脑自动调节 (CA) 引导的 CPP 进行个性化治疗与 TBI 患者的预后改善有关 。值得记住的是,结合脑组织供氧和 ICP/CCP 指导的治疗可显著着改善有利的长期结果 。此外,在最近的一项荟萃分析中,Xie 等人证明这种联合疗法对 TBI 后患者的死亡率、ICP/CPP 和住院时间没有任何影响 。
在氧分压 (PaO 2 )(75–100 mmHg;7–13.33 kPa)的生理范围内,只要 PaO 2 不低于 50 mmHg (6.67 kPa ),PaO 2对整体 CBF 几乎没有影响。这是因为 CBF 与动脉氧含量而非 PaO 2相关。血红蛋白-氧解离曲线的形式表明动脉氧含量在讨论的 PaO 2范围内相对稳定 。
在功能性神经激活(神经血管耦合)期间,脑血管张力变化和 CBF 增加的梯度独立机制可能会增强决定大脑中氧水平的主要梯度 。这种机制的主要作用是在神经元激活过程中在消耗增加之前运输更高水平的氧 。
脑损伤后脑灌注和代谢受损已被反复记录。脑损伤后的不良结果与灌注不足、葡萄糖代谢降低和 CMRO 2 有关。最近的数据记录了创伤性脑损伤后 CMRO 2和格拉斯哥昏迷评分 (GCS)之间的联系。Soustiel 等人证明在 TBI 患者中,CBF 在最初 24 小时内有所减少,并且在不良结果后观察到更严重的低血容量。重要的是,CMRO 2和脑葡萄糖代谢率 (CMRG) 的降低与较差的结果相关 。
耗氧量
氧通过血液从毛细血管扩散到线粒体被输送到脑细胞,直到它作为氧化代谢的一部分在线粒体中被消耗掉。CMRO 2是大脑和健康清醒人群的消耗和能量稳态速率,平均为 3.3 mL/100 g/min 。它与CBF有关。在代谢需求升高的情况下,脑血管系统扩张以提供 CBF 的适当增加。
重要的是,随着神经活动的增加,CMRO 2也会增加 。在正常、未受刺激的大脑中,能量主要由葡萄糖氧化提供。然而,氧糖比的代谢率 CMRO 2 /CMR(glc),称为氧糖指数 (OGI),在激活过程中会增加并偏离教科书值 6。此外,在感觉(如视觉)刺激期间,大脑中的乳酸水平会增加 。与糖酵解相比,这种氧化代谢产生更多的能量,但对该过程的精确测量是有限的 . 线粒体具有高代谢活性,在有氧能量产生中起着关键作用,其主要功能是通过氧化磷酸化产生三磷酸腺苷 (ATP)。
线粒体功能障碍是细胞损伤发生的主要因素。缺血/再灌注期间的成功复苏需要通过含氧血液的再灌注重建有氧代谢。线粒体作为再灌注损伤的效应器发挥着重要作用。细胞器的损伤会损害细胞质中细胞色素 c 的氧化磷酸化和消除。主要机制是氧化应激和 Ca 2+过载 。
氧输送的紊乱会停止电子流动并中断在上述 ATP 生产中很重要的“质子动力”的产生。当然,细胞可以通过糖酵解厌氧产生 ATP。然而,这个过程效率较低,不足以满足代谢需求,最终产品是乳酸盐。
细胞内的氧气
线粒体的作用是在氧气的生理范围内维持 ATP 的最大水平。重要的是要记住,还有其他机制负责耗氧量。大多数情况下,O 2被线粒体消耗,但 1–2% 的氧气未完全还原为超氧阴离子 (O2−)(图 1)。
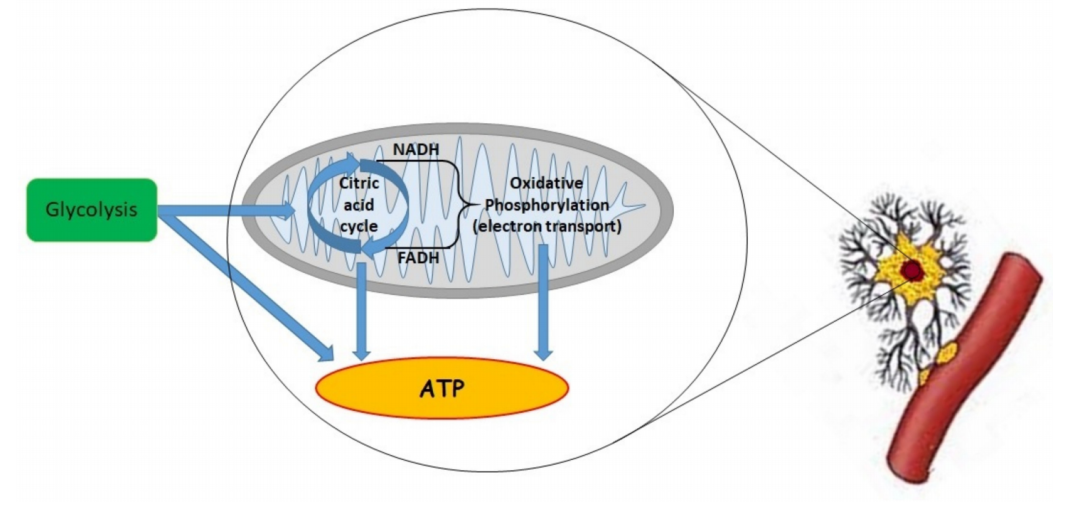
图 1.减少三羧酸 (TCA) 循环产生的 烟酰胺腺嘌呤二核苷酸 (NADH) 和黄素腺嘌呤二核苷酸 (FADH 2 ) 成分时的“线粒体呼吸”益处。在线粒体内膜中,NADH 和 FADH2 产生的电子被复合物 I 和 II 氧化为 NAD+ 和 FAD+。之后,这些电子依次转移到复合物 III、细胞色素 c 和复合物 IV。细胞色素 c 氧化酶(COX,复合物 IV)将电子传递给分子氧。这是连接氧与氧化磷酸化的线粒体电子传递链 (ETC) 中的一种重要酶 . 电子通过 ETC 的传输与从线粒体基质穿过内膜到线粒体间膜空间的质子传输有关。这种易位产生了质子的电化学梯度(pH 梯度和膜电位)。这些分子可能漂移通过 F1Fo-ATP 合酶(复合物 V)或回到线粒体基质 。重要的是,复杂的 V 连接质子转移到从二磷酸腺苷 (ADP) 和磷酸盐生产 ATP。在正常氧气水平下,丙酮酸作为糖酵解的产物,被输送到线粒体中,并通过丙酮酸脱氢酶 (PDH) 复合物转化为乙酰辅酶 A . 之后,乙酰辅酶 A 与草酰乙酸结合并产生柠檬酸盐——三羧酸循环的第一步。减少该循环中的当量会影响 ETC 产生 ATP 和用于信号传递的活性氧 (ROS),而 TCA 的中间体用于生物合成过程,例如脂质合成
生物体发展出重要的适应机制 。这些机制之一是代谢抑制。在体外氧水平在 1% 到 3% 之间时观察到细胞中线粒体耗氧量的减少。当氧气 (<0.3%) 水平开始限制细胞色素 c 氧化酶 (COX)(复合物 IV)时,就会出现“氧气一致性”. 由于氧气可及性有限,缺氧会导致氧化磷酸化减少和再合成的磷酸盐、ATP 和磷酸肌酸的损失。ATP 依赖性 Na/K 泵也发生变化,促进 Na、Ca 和水流入细胞,引起细胞毒性水肿。此外,局部缺血影响腺嘌呤核苷酸的分解代谢,导致细胞中次黄嘌呤的积累。细胞溶质钙升高促进多种途径,例如磷脂酶的激活,重要的是前列腺素、脂肪酶、蛋白酶和核酸内切酶的释放,这会破坏细胞的结构元素 . 此外,在内皮中促炎基因产物(白细胞粘附分子、细胞因子、内皮素凝血恶烷 A2)表达增加后,观察到促炎状态。相反,前列环素和一氧化氮被抑制。
换气过度/换气不足
影响脑灌注的最有力因素之一是通气过度/通气不足,对 CBF 和 PaCO 2有影响。过度通气是一种常用疗法,用于降低升高的 ICP 或放松紧张的大脑(低碳酸血症降低的 CBF 和 CBV)。在创伤性脑损伤 (TBI) 患者中,当 PaCO 2从 40 mmHg 降至 30 mmHg时,过度通气会导致 CBF 降低 34%,脑血容量 (CBV) 降低 9% 。然而,过度换气和低碳酸血症,除了血管收缩和 CBF 降低外,还会引起神经元兴奋性和癫痫发作持续时间延长,兴奋性氨基酸增加和脑脊液碱中毒,氧 - 血红蛋白解离曲线 (OHDC) 左移。所有这些机制都可能导致氧供应和输送的减少以及氧摄取的显著增加。
应该注意的是,二氧化碳是一种常见的分子,其生理范围为 35–45 mmHg。低碳酸血症(二氧化碳分压 <35 mmHg)和轻度高碳酸血症 (>45 mmHg) 会导致重要的神经系统紊乱。最近的数据表明,高碳酸血症具有神经保护机制,并可能通过脑血管舒张改善 CBF。高碳酸血症还会导致脑水肿、ICP 升高、氧合血红蛋白解离曲线右移、全身血管阻力 (SVR) 降低以及组织可用氧增加 。
过度通气是一把双刃剑,有一些短期的有益影响和长期的风险。ICP 正常的 TBI 患者的初始 PaCO 2值应在 38-42 mmHg 的正常范围内。TBI 患者机械通气期间的控制性过度通气(从不低于 30 mmHg 的 PaCO 2)是一种经批准的治疗性、临时(在受伤后的前 24 小时内)挽救严重颅内高压生命的干预措施 。然而,应使用多模式神经监测方法对每个患者的 PaCO 2水平进行调节和个体化。
影响 CRMO2 和细胞氧平衡的“4-H”因素
脑细胞特别容易受到缺血性损伤,因为它们的能量代谢、高代谢率、有限的内在能量储存以及与葡萄糖的有氧代谢的关键关系等几个不寻常的特征。因此,细胞代谢和关键化合物的消耗可以通过药物或临床状态改变,可以概括为“4-H”。
1. 缺氧(Hypoxia)
大脑是对缺氧、复氧和氧化应激最敏感的器官之一。如上所述,大脑对代谢氧的需求非常高,并且极易受到缺氧损伤(表 1)。
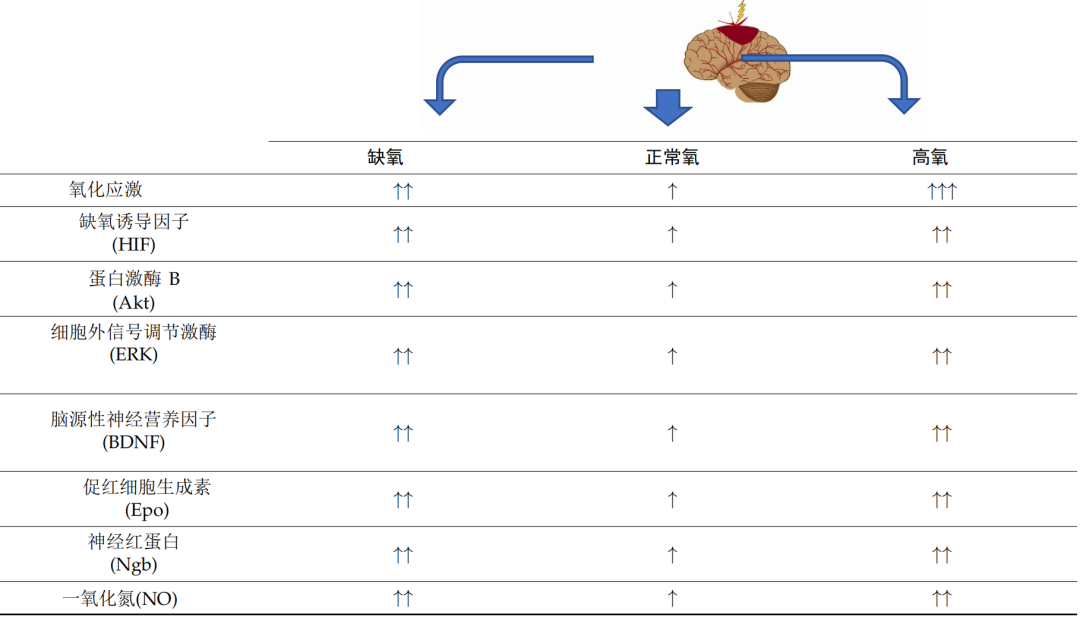
表 1. 大脑供氧障碍对选定通路和因素的潜在影响。向上箭头表示机制的方向,可能会在不同程度上得到强化,具体取决于致病因素。箭头的数量定义了过程的强度。
线粒体中的氧化应激发生在氧化还原失衡的状态下,并且积累了超氧自由基、羟基自由基或一氧化氮自由基等小的氧化剂模式。需要注意的是,缺氧和高氧都可能诱导氧化应激和细胞凋亡 . 急性缺氧会增加大脑中 ROS 的产生,而复氧会促进这一过程。低氧水平会导致脂质过氧化、蛋白质氧化和一氧化氮水平以及抗氧化防御系统增加。超氧化物歧化酶 (SOD)、还原型谷胱甘肽 (GSH)、谷胱甘肽过氧化物酶 (GPx) 和还原型/氧化型谷胱甘肽 (GSH/GSSG) 比率) 在脑细胞中受到显著抑制。在缺氧状态下氧稳态和血管生成控制的关键调节剂之一是 HIF(缺氧诱导因子)。存在三种转录因子,HIF-1、HIF-2 和 HIF-3 . 这些异二聚体由β亚基(HIF-1β、HIF-2β和HIF-3β)表达,并与α亚基HIF-1α、HIF-2α和HIF-3α相连,直接影响缺氧。HIF-1 和 HIF-2 是具有独特靶基因的转录调节因子。HIF-1 调节对缺氧的急性反应(<24 小时),由 HIF-1 形成的网络倾向于增加灌注和增加氧气水平 。
只要PaO2不低于50 mmHg,对全脑球CBF的影响就不大。在这一点上,随着PaO2的进一步恶化,血流会急剧增加。缺氧时ATP水平的降低打开了平滑肌上的KATP通道,导致低极化和血管扩张. 重要的是,缺氧会进一步降低PaO2,而CBF可能增加到基线水平的400%。CBF的改变不影响新陈代谢,但血红蛋白饱和度从100%(当PaO2>70 mmHg(PaO2>9.33 kPa)时)下降到50%(当<50 mmHg(<6.66 kPa)时)。此外,PaO2的降低会增加局部NO和腺苷的产生。慢性缺氧通过影响毛细血管密度增加CBF . 能量细胞衰竭和延迟凋亡与NO-有关,由乳酸中毒刺激一氧化氮合酶(nNOS)和离子转运的破坏所催化。
此外,神经元膜转化导致谷氨酸释放,从而促进 N-甲基-D-天冬氨酸 (NMDA) 受体的激活和钙内流,促进脂肪酶、蛋白酶和内切核酸酶,从而促进自由基形成[。最后,观察到炎症、严重的线粒体功能障碍和 ROS(超氧化物、羟基、过氧化氢等)的产生以及脂质、蛋白质、细胞和脱氧核糖核酸 (DNA) 的氧化。
在动物模型中,氧化应激参数和抗氧化系统在脑损伤后 24 小时恢复到控制系统 。Coimbra-Costa 等。记录表明,在复氧 24 小时后,氧化应激减少,但细胞凋亡得以保留,尤其是在海马体中 。海马体细胞凋亡率高于皮层可能是缺氧性脑损伤脑功能受损的原因。缺氧状态下氧稳态和血管生成的关键调节因子之一是缺氧诱导因子 (HIF) . HIF-1 与细胞核中基因启动子上的缺氧反应元件 (HRE) 结合,并促进靶基因的转录,如血管内皮生长因子 (VEGF)、葡萄糖转运蛋白 1 (GLUT1) 和其他如糖酵解酶、乳酸脱氢酶或促红细胞生成素。HIF-1 在星形胶质细胞和雪旺氏细胞的糖酵解上调中也至关重要 . 相比之下,HIF-2 和 HIF-3 的表达在内皮细胞中是在慢性缺氧条件下开始的。重要的是,在内皮适应长期缺氧的过程中观察到从 HIF-1 到 HIF-2 和 HIF-3 的转换。HIF-1 通过形成初级和非常原始的网络来覆盖血管生成,随后 HIF-2 和 HIF-3 的表达稳定并促进该脉管系统的成熟 。此外,由 HIF-1 格式化的网络容易导致灌注升高和氧水平升高。
慢性缺氧下促进蛋白酶体降解的另一种机制是基于 Hsp70 相互作用蛋白(Hsp70/CHIP 复合物)的羧基末端 。活化激酶 C1 (RACK1) 的受体还导致 HIF-1α 的降解和热休克蛋白 90 (Hsp90) 的促进,从而保护 α 亚基。此外,RACK1 会导致 HIF-1α 的蛋白酶体降解和泛素化 。此外,Kruppel 样因子 2 (KLF2) 在内皮细胞中表达并负责生理性血管形成,通过中断 Hsp90 连接以“von Hippel-Lindau 独立但蛋白酶体依赖性方式”激活 HIF-1 缺氧降解与 HIF-1 。
微小 RNA (miRNA) 是一个具有 18-22 个核苷酸的非编码 RNA 分子家族 。人们越来越关注 miRNA 在中枢神经系统发育和功能中的关键作用,作为“切割和沉默基因表达”的基因调节剂 。相比之下,在各种神经系统疾病中记录了异常水平的 miRNA 。miRNA 210 主要在缺氧状态下表达,由 HIF1 α 促进,并在缺氧缺血损伤中建立神经保护作用。
miRNA 分子通过抑制半胱天冬酶[来减少神经元细胞的凋亡过程。因此,随着人们对 miRNA 模式与缺氧/缺血的关联越来越感兴趣,这些分子可能是缺血的临床生物标志物和单独的 miRNA 治疗复合物 。最近已显示糖皮质激素 (GCs) 在缺氧和缺血/再灌注条件下的保护作用 。GC 给药导致中枢神经系统对缺氧的耐受性增加 。急性缺氧会激活下丘脑-垂体-肾上腺 (HPA),并在血清中积累长达 24 小时的皮质酮 . 最近的数据表明,低氧耐受性与 HIF-1 α 的上调和 GC 的释放增加有关。GC 受体和 HIF-1 之间的直接串扰可能是 GC 上调 HIF-1 靶基因的生化途径的基础 。
缺氧的临床意义:
-
脑组织氧合作用减少是严重创伤性脑损伤后不良结果的预测指标。
-
缺氧缺血性脑损伤 (HIBI) 与显著的死亡率和发病率相关 。
-
LOCO 2研究表明,以较低的 PaO 2为目标可改善急性呼吸窘迫综合征 (ARDS) 患者的预后 。
-
脑组织氧张力 (PbtO2 )至关重要,它是继 ICP 之后的第二个监测变量,代表了 TBI 患者的多模态监测。
-
继发性缺氧与 CSF 中细胞因子的持续产生以及髓鞘碱性蛋白 (MBP) 和 S100 等血清生物标志物的过度升高有关 。
-
MBP、S100 和神经元特异性烯醇化酶 (NSE) 生物标志物在缺氧和不利结果的患者中更高(扩展格拉斯哥结果昏迷评分 (GOSE) 1–4)
-
HIBI 作为二次打击模型,是原发性和继发性缺血/缺氧损伤导致神经血管单元整体毁灭性严重损伤的结果
-
继发性脑缺氧与新生神经元和星形神经胶质细胞损伤有关。重要的是,继发性缺氧与脑促炎反应有关,但与平行的脑内皮损伤无关。
-
基于 PbtO 2和 ICP 监测的方案可显著缩短 TBI 后的脑缺氧时间 。
-
急性间歇性缺氧 (AIH) 和特定任务训练 (TST) 可协同改善中枢神经系统损伤后的运动功能 。
2. 高氧(Hyperoxia)
高氧毒性的概念是由 ROS 的内源性产生定义的。
100%氧疗后线粒体结构的实验检查显示线粒体肿胀巨大,线粒体膜和嵴被稀释和损坏,这与皮质脑中的髓鞘、轴突和细胞器损伤直接相关。高氧与 Akt 表达和/或磷酸化的抑制有关,这是低氧水平的逆转。实验研究表明,在大鼠高氧模型 (FiO 2 0.4–0.8 ) 中,p-Akt 表达稳定下降,随着时间的推移直至 12 小时,然后逆转至基线值 。因此,Akt 信号在缺氧时增加,在高氧时抑制 .
丝裂原活化蛋白激酶 (MAPK) 参与 PI3K-Akt 信号通路,这是一条对缺氧或氧化应激具有神经保护作用的重要通路 。大鼠模型的最新数据表明,高氧 (FiO 2 0.4–0.8) 会降低 p-ERK1/2 激活直至 12 小时,然后在随后的 12 小时内恢复 。此外,大鼠脑模型中的高氧会影响 BDNF 和神经营养因子 3 和 4 的下调,并在随后的 15-20 小时内进行校正,从而导致高氧相关的凋亡性神经变性。在不同脑区观察到的促红细胞生成素受体 (EpoR) 结合在氧代谢中也起着重要作用. 在缺氧条件下,它会上调,因为 HIF-1α 与 Epo 结合,显示出与缺血缺氧/复氧损伤相反的神经保护作用。实验数据表明,在用 FiO 2 0.5 处理 3 周的小鼠中,高氧会上调 Epo,升高 HIF-2α,但在 FiO 2 = 0.3处理的 4 周期间,只有 EpoR 表达增加。值得注意的是 NO,它通过改善微血管系统中的脑血流来改善氧输送 。此外,NOS 通过改善血管自动调节呈现神经保护作用. 它触发不同的机制,例如 BDNF 表达、HIF 稳定化、HIF 的 S-亚硝基化、阻断 HIF-1α 降解、与 MAPK 和磷酸肌醇 3-激酶 (PI3K) 信号相互作用以及EpoR表达上调。在呈现所选 NOS 过度活跃的非生理状态下,NO 开始作为自由基具有神经毒性。高氧浓度控制 NOS 表达并通过过量释放超氧阴离子抑制 NO 血管舒张和促进大脑血管收缩来抑制 NO. 重要的是,超氧阴离子与 NO 的连接会促进具有破坏性的过氧亚硝酸盐 (ONOO−)的产生。在动物研究中,NO 通过环氧合酶 2 和前列腺素 E2 抑制与高氧诱导的星形胶质细胞增殖和促炎反应有关。
高氧的临床意义:
-
高氧与更高的死亡率和更差的短期功能结果相关,尤其是在脑损伤后的前 24 小时内接受不受控制的氧输送的患者(可能是因为高氧诱导的氧自由基毒性伴或不伴血管收缩).
-
高氧浓度的潜在毒性(接受超过 0.6 的FiO 2的患者)。
-
先前的研究表明,较高的吸入氧浓度与急性肺损伤相关,伴有轻度至重度弥漫性肺泡损伤 (DAD) 。
-
动脉瘤破裂后 72 小时内的高氧水平是脑血管痉挛的一个未受影响的预测指标 。
-
此外,与保守治疗相比,自由氧疗增加了 30 天死亡率 。
-
世界卫生组织关于预防手术部位感染的全球指南对减少手术部位感染 (SSI) 的高剂量氧疗建议存在争议。
-
高氧血症可能会降低心血管衰竭患者的心输出量并增加全身血管阻力 。
3. 高热(Hyperthermia)
由于下丘脑直接损伤、脑部炎症或继发感染导致发热而导致脑损伤后的患者经常观察到体温升高。
全身性高热在脑损伤后很常见。在脑损伤患者中,它与不良的神经学结果有关,因为它容易导致更严重的继发性损伤 (图 2)。
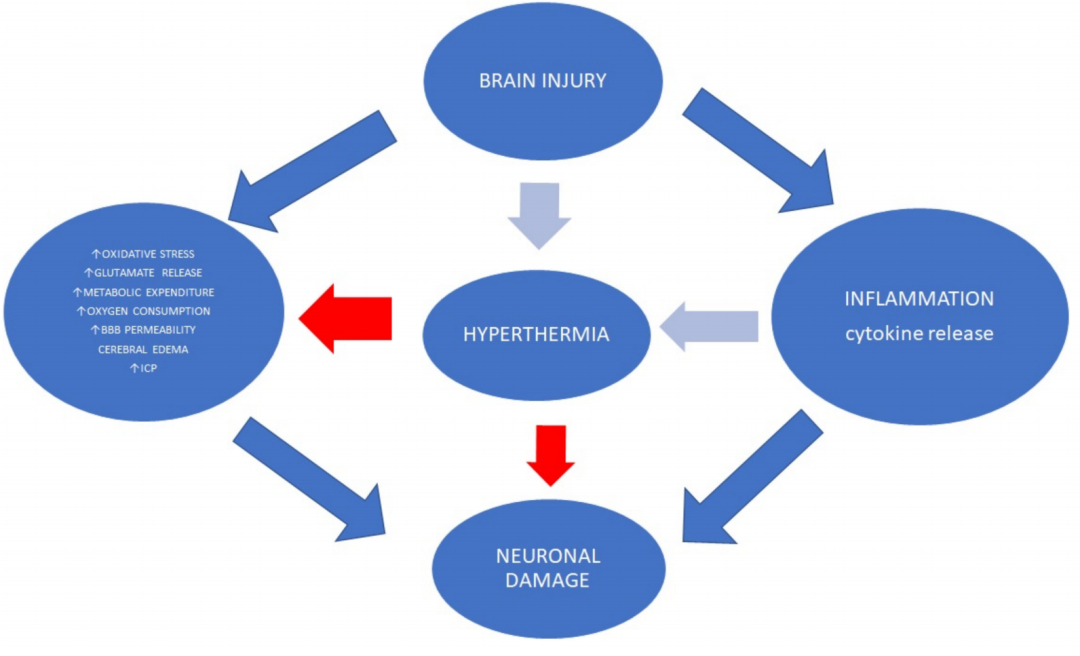
图 2. 体温过高与神经系统预后不良有关,因为它容易造成更大的继发性损伤。温度变化导致细胞因子释放升高、中性粒细胞活性升高和代谢消耗升高。高热疗还会增加 ROS 的产生和细胞凋亡。
应该注意的是,体温过高并不总是与发烧有关。发烧是一种适应性反应,例如中性粒细胞迁移增加、T 淋巴细胞激活以及白细胞介素 1 和干扰素生成增加。温度变化导致细胞因子释放增加、中性粒细胞活性增加和代谢消耗增加、白细胞积聚增加、血管通透性增加和轴突损伤 . 温度变化也会导致脑血流转换,从而引起脑氧合作用的变化。最近的动物研究表明,热疗与 CD18 和细胞间粘附分子 1 (ICAM-1) 的激活以及皮层中离子化钙结合衔接蛋白 1 (IBA-1) 反应性小胶质细胞的增加有关 。高热还会增加 ROS 的产生和细胞凋亡,例如通过 c-Jun N 末端激酶 (JNK) 激活。Wettervik 等人报道,体温过高会导致能量代谢紊乱,与较高的 ICP 和较低的 CPP 无关 . 重要的是,较高的温度与脑组织中较低的葡萄糖浓度以及 5 天后乳酸 - 丙酮酸比率 > 25 的较高百分比有关 。此外,作者并未表明体温过高与较差的临床结果之间存在联系。
在基线核心体温增加超过 1.5–2 °C 期间,代谢率增加约 20–25% 。最近的数据表明,核心体温升高会影响大脑葡萄糖利用率和 CMRO 2每摄氏度增加 5% 至 10% 。Nunneley 等人观察到体温升高超过 2°C 与下丘脑、丘脑、胼胝体、扣带回和小脑的葡萄糖代谢率较高以及尾状核、壳核、脑岛和后扣带回的葡萄糖代谢率较低有关 。此外,体温升高 2°C 后大脑新陈代谢增加 10% 可能与支持氧合作用的血流量显著减少有关 . 此外,Spiotta 等人证明高热不会减少脑组织的氧。应该注意的是,较高的体温导致更好的维持 O 2摄取 (VO 2 ) 的能力,因为在 O 2供应开始之前摄取了更高比例的输送 O 2 。体温过高期间心血管调节以及交感神经活动也会影响 CMRO 2和 CBF 之间的耦合。交感神经的活动在高温下增加。肾上腺素能神经围绕着血管系统,尤其是脑动脉 . 一些作者认为,高热下的血管收缩会导致 CBF 降低 。但是,有一些疑问。首先,在高温下的高代谢状态下,不同的药物,如组胺、一氧化氮或前列腺素可能会抵消血管收缩 。其次,血压显著影响脑血管系统。第三,脑血管的异质性反应可能会因体温过高以及 α 和 β 肾上腺素能受体密度的变化而改变 。体温升高会损害血脑屏障 (BBB) 的完整性,尤其是在脱水时。最后,Bein 等人记录了 PaCO2 正常化至正常碳酸血症导致 CBF 恢复到生理状态。
高热的临床意义:
-
脑损伤后早期常发生发热等全身并发症,加重继发性脑损伤 。
-
高达 50% 的急性脑损伤患者在住院期间会出现发热 。
-
大脑温度变化 (>1 °C) 与不良功能结果相关 。
-
总之,较高的体温与代谢需求增加和内源性应激水平升高、血压水平变化、心输出量和心率增加、换气过度、兴奋性氨基酸的突触释放、ICP 水平升高、缺血性皮质去极化和血脑屏障故障有关。
-
没有氧输送不匹配的高热似乎不会引起显著的神经化学改变,例如葡萄糖、乳酸、丙酮酸和谷氨酸水平 ]。
-
PbtO 2可能是在高体温发生期间监测的重要元素,以提供对大脑中氧代谢的看法。
-
在严重创伤性脑损伤患者温度升高的情况下,可以观察到PbtO2的变化。PbtO2平均每隔三分之一就会上升,每隔六次高温就会下降。最近的数据表明,PbtO2的斜坡可能与CPP和MAP的降低同时发生
-
防止发烧的体温管理对于严重创伤性脑损伤患者至关重要。严重脑损伤的国际指南强调了核心温度测量和高于 38°C 治疗的重要性 。
4. 低温(Hypothermia)
当前国际临床指南的主要目标是通过抑制继发性损伤来改善最终结果,尤其是在损伤后的急性期。这些协议还包括校正温度和治疗性低温。中度至深度低温可抑制炎症并减少兴奋性毒性和自由基的产生,这是神经保护机制之一。不同程度的低温可提高神经元对缺血的耐受性并抑制神经元死亡]。脑低温可降低ICP、维持BBB 功能并改善葡萄糖利用. 此外,较低的温度会抑制缺氧脑去极化,释放神经递质,并通过蛋白酶激活和高能量磷酸盐消耗率降低新陈代谢。作者甚至指出,深低温会影响大脑 ATP 的产生,并将心脏骤停后的存活率提高三到四倍。重要的是,缺血期间的低温可减少脂质过氧化并从根本上减少 ROS 的产生。低温可降低 JNK 激活和细胞凋亡率 。低温还会激活一系列神经炎症,并可能将 M1/M2 巨噬细胞极化改善为有利的表型 .
较低的温度可改善 TBI 和脑缺血后的脑代谢。在动物模型中,葡萄糖 (CMR glc ) 和 CMRO 2的代谢率降低,但显着的是,ATP 分布的降低超过了合成的降低。此外,在含氧量正常的状态下,低体温会降低大脑的耗氧量以及 CBF 和氧输送的附带消耗(脑血管阻力升高和大脑中氧摄取的微量变化)。适度缺氧导致 CBF 和氧摄取升高,随后脑血管阻力降低 . Chihara 等人在大脑温度降低 1.6° ± 0.1° 和缺氧的动物模型中记录了低体温导致氧输送、耗氧量和 CBF 减少。此外,观察到脑血管阻力显著改善,并且没有氧摄取转移 。Hashem 等人最近的数据,使用近红外光谱 (NIRS) 和磁共振成像 (MRI) 方法,分别显示大约 37% 和 32% 的低温小鼠和大鼠的皮质中CMRO 2显著减少 . 因此,通过 NIRS 装置等不同方法靶向脑组织氧合可能是脑损伤治疗指南的一个重要方面,用于改善脑氧合、监测脑血管反应性 (CVR) 和最终结果]。
低温的临床意义
-
低温治疗是当前临床实践指南的重要组成部分。
-
治疗性低温使用不同的冷却方法将大脑温度维持在目标水平。
-
低温治疗可改善神经系统结局。相比之下,TBI 后入院时意外体温过低会导致更高的住院死亡率 。
-
最近发表的数据并未提倡在 TBI 患者损伤后的前 6 小时内进行早期预防性低温治疗 。
-
TBI 后 35 至 35.5 °C 的体温可降低颅内高压并保持足够的 CPP,而不会出现心功能不全和氧债 。此外,低温可降低高 ICP。
-
最近的荟萃分析记录了体温测量在院前管理中避免体温过低的重要性 。
未来疗法
所讨论的与氧相关的机制已成为脑损伤治疗的目标。管理各种形式脑损伤患者的一个关键因素是维持氧稳态、供应和消耗,转化为正常的线粒体代谢。缺氧和高氧都可能对最终的神经学结果产生负面影响。最近的研究结果表明氧疗在与常压高氧 (NBHO) 相关的神经保护中的作用。接受高氧水平 (FiO 2 0.6–1.0) 治疗两个小时的急性脑损伤患者表现出改善的氧化还原平衡和降低的乳酸/丙酮酸比率 (ΔLPR -3.07 p = 0.015) 。 NBHO 方法基于在正常大气压下连续供氧。实验数据已证明 NBHO 在缺血性中风、出血性中风和脑外伤中的益处。
杨等在动物模型中,实验记录了常压氧疗 (60%) 对神经功能、水肿和 HIF-1α、水通道蛋白 4 (AQP4) 和 Na+/H+ 交换器 1 (NHE1) 表达的影响(分别为p < 0.05)。这些作者表明,治疗会抑制 NHE1 表达和 Na+ 内流。这些作用导致水通过 AQP4 运动后的脑水肿减少。高压氧疗法 (HBOT) 是 TBI 中提出的另一种疗法。HBOT是在大于1个绝对大气压的压力下100%吸氧。HBOT 抑制炎症并保护 BBB 完整性并支持血管生成和神经生成. 动物模型的最新数据表明,在脑损伤后的早期进行氧疗可显著降低 NF -κB 和细胞外组蛋白 H1、H2A 和 H4 的表达。组蛋白是细胞核中的结构蛋白,是缺氧和缺血引起炎症的重要因素。此外,HBOT 可抑制神经元细胞的凋亡机制并保留线粒体膜的特性,从而减少继发性损伤。当然,HBOT的临床疗效仍有争议。罗克斯沃尔德等人。一项小型研究表明,HBOT 并未改善一组闭合性颅脑损伤患者的预后 . 然而,在 2013 年的其他 II 期临床试验中,Roackswold 等人。证明联合 HBOT 与常压高氧 (NBHT) 疗法可改善氧化代谢和氧脑组织分压水平。此外,该疗法还降低了颅内高压、死亡率并改善了结局(通过 GOSE 测量)。另一项研究表明,HBOT 可显著改善创伤后应激障碍症状、认知功能并减少抑郁和焦虑。
HBOT 的争议性影响可以用高氧-低氧悖论 (HHP) 来解释。最近的研究表明,重复和周期性高氧可能会诱发分子机制并激活类似于缺氧的介质。在间歇性高氧期间观察到 HIF、VEGF、SIRT、线粒体生物发生和干细胞增殖的激活。
另一种治疗选择是乳酸在脑能量代谢中的重要作用 ]。缺血性脑损伤中的实验性乳酸补充剂会影响谷氨酸和γ-氨基丁酸 (GABA) 的释放,并改善脑电图 (EEG) 。此外,Berthet 等人证明补充乳酸可抑制氧气和葡萄糖输送障碍中的神经元死亡 。在大脑中动脉闭塞和缺血模型中进行的相同治疗也表现出显著的神经保护作用 . Ichai 等人在随机对照试验中指出,在一组 TBI 患者中,高渗乳酸钠 (HSL) 治疗在降低升高的 ICP 方面比甘露醇更有效]。此外,这种效果持续时间更长,并且与颈静脉 O 2饱和度、血浆中的葡萄糖和乳酸水平以及 pH 值的改善有关。患者还表现出更好的神经学最终结果 。输注 HSL 3 小时会影响细胞外代谢物。一种理论认为这些溶液含有可代谢的乳酸盐和钠离子。大脑中的乳酸会引起阴离子和正电荷之间的不平衡,并通过补偿阴离子流出来抵消有害的细胞肿胀 。
最近的数据显示大脑中乳酸、丙酮酸和葡萄糖水平升高,与谷氨酸和 PbtO 2值以及 ICP 相关联。布扎特等人报道,这些影响可能是大脑代谢转变为乳酸利用率升高的结果,从而避免了葡萄糖的影响。此外,脑氧合作用的抑制可能继发于碱中毒,碱中毒会增加氧气对血红蛋白的亲和力,并提示有益效果。总之,高渗乳酸钠输注可降低谷氨酸相关的兴奋性毒性、改善脑灌注、缓冲代谢性酸中毒、减轻脑水肿和 ICP 并改善心脏功能。
结论
氧对脑细胞的功能至关重要。因此,导致氧气供应和消耗中断的机制是持续深入研究的主题。越来越需要新的治疗方法来减少病理细胞过程的级联。
原文:Siwicka-Gieroba. Cerebral Oxygen Delivery and Consumption in Brain-Injured Patients. J Pers Med 2022;12:1763

