王共强:肝豆状核变性螯合治疗与铜缺乏
时间:2023-10-08 11:19:49 热度:37.1℃ 作者:网络
论坛导读:铜是一种必需的微量金属,是催化几种重要的细胞酶所必需的。然而,由于过量的铜可能催化有毒活性氧物质的产生,因此铜的转运和细胞铜含量受到严格的控制。铜缺乏和过量已经被认为是世界范围内婴儿和儿童的潜在健康问题。铜缺乏和过量的临床表现已得到很好的描述,但高铜摄入与遗传控制系统相互作用的确切顺序尚不清楚,从而导致婴儿肝脏损伤。与铜稳态的功能发育相关的基因突变或表观遗传因素可能会使正常摄入铜的正常婴儿更容易受到铜中毒的影响,这一直是一个令人关注的问题。
肝豆状核变性,也称为Wilson病 (Wilson's disease ,WD),是一种遗传性铜代谢障碍,伴有全身性铜超载,导致肝脏和神经精神等临床症状。WD的发病机制包括跨膜铜转运型P ATPase 7B的功能障碍,其参与铜从肝细胞向胆汁的排泄和铜向铜蓝蛋白(Cp)的掺入。这种转运蛋白的失效导致铜在肝细胞中的进行性累积,肝细胞的进行性损伤和生物可利用的所谓“游离”非Cp结合铜(NCC)释放到血液中,伴随二次铜累积,以及对其他器官和组织的损伤。在WD的病程中,观察到高血清NCC,通常高于15 g/dL(正常范围:5-15g/dL),与此相反,血清总铜随着血清Cp水平的降低而降低至70 g/dL以下(正常范围:70-140g/dL)。由于WD是由铜体超负荷和中毒引起的,所以药物治疗是基于药物,导致全身铜平衡为负。螯合剂(d-青霉胺[DPA]和trientine [TN])增加尿中铜的排泄,而锌盐(ZS)减少消化道对铜的吸收,目前在临床试验中,钼盐(四硫代钼酸盐[TH])已显示螯合过量的铜,从而通过胆汁排泄减少铜水平,并减少消化道对铜的吸收。
人们越来越认识到铜是动物和人类都需要的重要矿物质。它在细胞呼吸、铁氧化和血红蛋白合成等许多代谢过程中起着至关重要的作用。铜缺乏可以是遗传性的或获得性的,可以导致多种疾病过程,如环状铁粒幼细胞性贫血、骨髓发育不良和全血细胞减少症。铜缺乏作为过度治疗的一种可能表现无疑是有吸引力的。同样矛盾的是这种缺乏发生在接受锌治疗的患者中,而不是接受螯合剂治疗的患者中,螯合剂在去除机体铜方面具有更强的作用。研究和文献综述显示肝豆状核变性患者治疗诱发的铜缺乏似乎是一种罕见的事件,但是,治疗肝豆状核变性的临床医生必须记住这一点,因为铜缺乏的临床和实验室体征可能会被忽略,并且可能会被错误地归因于其他原因。此外,与铜缺乏引起的血细胞减少不同,临床上铜缺乏引起的神经系统症状(尤其是脊髓受累的症状)倾向于持续存在,尽管铜缺乏进行了治疗。因此,在肝豆状核变性患者的治疗中,非常需要避免这种不良事件。
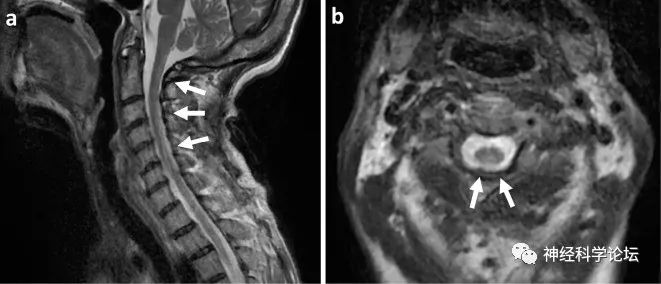
颈髓的T2加权磁共振成像。矢状面(a)和轴面(b)显示从C1到C6的脊柱纵向高信号损伤。图源:Intern Med. 2023 Apr 1;62(7)
铜缺乏症是肝豆状核变性长期治疗的一种罕见并发症,通常伴有高血清锌水平。铜缺乏可以被认为是对去铜效果的夸大,这是肝豆状核变性药物治疗的目标。要解决的第一个问题是用目前的疗法为肝豆状核变性患者去除铜的实际可能性。在现实世界中,如果我们考虑到在一系列肝活检中评估肝铜浓度的研究表明铜保持升高,即使在临床进展良好的患者中,也很难获得真正的铜耗竭。因此,这两种现象是如何共存的:一方面,肝豆状核变性患者的肝脏中持续的铜积累得到了良好的治疗,另一方面,这种治疗在他们中的一小部分患者中引起异常铜缺乏的风险?
由于目前很少对肝豆状核变性患者进行肝活检,无论是在诊断时还是在随访期间,目前还不清楚治疗引起的铜缺乏肝豆状核变性患者的肝铜蓄积情况。鉴于铜缺乏的现象主要在锌治疗期间观察到,锌治疗的主要作用机制是通过与金属硫蛋白结合而减少肠道铜吸收和稳定肝脏铜,从而减少游离毒性部分,可以假设,即使在铜缺乏的情况下,肝脏中仍存在一定量的铜积累。在这种情况下,了解铜缺乏患者Kayser‐Fleischer环的变化肯定是有用的,因为在治疗良好的肝豆状核变性患者中Kayser‐Fleischer环消失是有据可查的。
铜是脑细胞的必需元素,因为它是参与能量代谢途径的各种酶的辅因子和结构成分。越来越多的证据表明,铜缺乏在铜稳态受损导致的神经变性中起着关键作用。尽管铜在线粒体呼吸中的作用无可争议,但其在脑组织中的稳态调节仍不清楚。对编码能量代谢关键途径的基因表达变化的评估可以极大地有益于探索铜在神经变性中的作用的进一步研究。
鉴于肝脏在维持铜代谢中的中心作用,肝硬化中存在铜缺乏就不足为奇了。在临床营养学中,铜缺乏的最佳定义是其血清或血浆浓度。确定的原因包括锌摄入过多、门克斯病、胃肠疾病或导致吸收不良和肾脏疾病的手术。因为没有一个铜缺乏的病人有这些症状,肝硬化可能是一个独立的和以前被低估的铜缺乏的危险因素。此外,铜缺乏不仅仅是蛋白质营养不良的附带现象,而是具有不同功能后果的独特微量营养素缺乏。循环铜和铜蓝蛋白之间的强相关性证实了这一观点,铜蓝蛋白的周转率取决于铜的利用率。
铜缺乏主要表现为血细胞减少,影响三个血系中的一个或多个,以及磁共振成像上的脊髓改变。不幸的是,血细胞减少在肝豆状核变性患者中几乎很常见,这可能是由于铜缺乏的作用受到质疑的多种因素,但通过治疗调节和铜补充的逆转有效地加强了铜缺乏的致病作用。此外,考虑到铜在造血过程中的作用(铜蓝蛋白和铁血红素是铁的输出和吸收以及血红素合成所需的铜依赖性铁氧化酶)以及镉在缩短红细胞寿命方面的影响,镉会导致血细胞计数异常也就不足为奇了。不太容易解释的是为什么铜缺乏只发生在一小部分病例中。在一项研究中,一名铜缺乏患者因不明原因接受了比通常推荐剂量高得多的硫酸锌,但在其他情况下,锌剂量是足够的。然而,不幸的是,铜缺乏的任何预测因素都已经被确定。口服锌诱导肠上皮细胞产生金属硫蛋白。金属硫蛋白通过与食物中的铜结合并排泄到粪便中来抑制肠道中过量的铜吸收。锌剂量不当导致金属硫蛋白过度表达,引起锌诱导性铜缺乏。当然锌治疗不是铜缺乏的唯一原因,还有其他一些因素也起作用。
有研究也提出了如何监测接受治疗的肝豆状核变性患者体内铜和锌沉积状态的问题。普遍认为在锌治疗期间,尿锌水平必须高于2000 μg (61 μmol)/24小时,而尿铜水平必须低于75 μg (1.2 μmol)/24小时。然而,最大尿锌阈值并没有显示,这可能是过度治疗的指示。最近,越来越多的注意力集中在铜尿的最低水平,低于这个水平就不能避免铜缺乏。在最近的美国肝病研究协会指南中,据报道出现血细胞减少、血清铁蛋白增加和/或24小时尿铜排泄不成比例地低(螯合治疗< 100 μg/24小时;< 20 μg/24小时[对于锌治疗< 0.3 μmol/24小时]可能表示过度治疗。研究和文献综述中缺乏关于铁蛋白行为的信息。这项研究再次强调了游离铜测定在监测接受治疗的肝豆状核变性患者中的作用,但众所周知,尽管最初的研究令人鼓舞,但它不是一项常规使用的测试。
因此,铜缺乏的风险增加了锌治疗的其他可能的副作用,如胃肠疾病和胰腺高酶血症。可能具有更严重副作用 (神经症状的反常恶化、过敏反应、骨髓抑制、神经病变等) 的螯合剂似乎不会导致铜缺乏。总之,尽管现有的药物在肝豆状核变性的治疗中取得了巨大的成功,但是不良反应的风险也不容忽视,因此,治疗肝豆状核变性患者的医生应了解并熟悉铜缺乏症状,主要包括贫血伴中性粒细胞减少和脊髓神经病变。铜缺乏的诊断主要基于临床症状和铜代谢,尤其是每日尿铜排泄量低。铜缺乏通常发生在长期接受治疗的患者身上,尤其是锌盐患者。铜缺乏的早期检测和暂时抗铜治疗停止(或剂量减少)导致血液学参数的快速改善。神经症状可能会在较长的时间窗内消退,但在长期铜缺乏的情况下可能会持续。
参考文献
Iorio R, Di Dato F, Spagnuolo MI. The Paradox of Copper Deficiency in Wilson's Disease Patients. Mov Disord Clin Pract. 2023 Jun 30;10(9):1304-1305. doi: 10.1002/mdc3.13817.
Chevalier K, Obadia MA, Djebrani‐Oussedik N, , et al. Can patients with Wilson's disease develop copper deficiency? Mov Disord Clin Pract 2023:1–11. 10.1002/mdc3.13813.
Sini M, Sorbello O, Sanna F, et al. Histologic evolution and long‐term outcome of Wilson's disease: results of a single‐center experience. Eur J Gastroenterol Hepatol 2013;25(1):111–117. 10.1097/MEG.0b013e328358f7da.
Araya M, Koletzko B, Uauy R. Copper deficiency and excess in infancy: developing a research agenda. J Pediatr Gastroenterol Nutr. 2003 Oct;37(4):422-9. doi: 10.1097/00005176-200310000-00005.
Cope‐Yokoyama S, Finegold MJ, Sturniolo GC, Kim K, Mescoli C, Rugge M, Medici V. Wilson disease: histopathological correlations with treatment on follow‐up liver biopsies. World J Gastroenterol 2010;16(12):1487–1494. 10.3748/wjg.v16.i12.1487.
Alkhouri N, Gonzalez‐Peralta RP, Medici V. Wilson disease: a summary of the updated AASLD practice guidance. Hepatol Commun 2023;7:e0150.
Cendrowska-Pinkosz M, Ostrowska-Lesko M, Ognik K, et al. Dietary Copper Deficiency Leads to Changes in Gene Expression Indicating an Increased Demand for NADH in the Prefrontal Cortex of the Rat's Brain. Int J Mol Sci. 2022 Jun 16;23(12):6706. doi: 10.3390/ijms23126706.
Nagral A, Sarma MS, Matthai J, et al. Wilson's disease: clinical practice guidelines of the Indian national association for study of the liver, the Indian society of pediatric gastroenterology, hepatology and nutrition, and the movement disorders society of India. J Clin Exp Hepatol 2019;9(1):74–98. 10.1016/j.jceh.2018.08.009.

