读书报告 | BRAFi和MEKi联合PD-1抗体治疗BRAF 突变型CRC的II期临床研究
时间:2023-11-26 22:26:32 热度:37.1℃ 作者:网络
导读
BRAF V600E突变发生在约10%的结直肠癌(CRC)中,驱动MAPK信号通路的持续激活。携带BRAFV600E突变CRC患者预后不良,对标准治疗反应较差,中位总生存期(OS)仅为BRAF野生型患者的一半。vemurafenib和dabrafenib等BRAF抑制剂对BRAFV600E突变黑色素瘤的有效率可达60%-80%,而BRAFi单药治疗BRAFV600E CRC的有效率仅为0-5%。既往研究指出,在BRAF被抑制后强大的自适应反馈网络可以迅速重新活化MAPK信号通路。因此基于BRAFi联合多种靶向MAPK反馈通路药物的临床研究被设计进行,以期提高BRAFV600E结直肠癌患者的应答率。
临床前研究表明,联合MAPK抑制和ICB可增强BRAF和KRAS突变型肿瘤的疗效,其潜在的协同作用机制包括BRAFi和/或MEKi对非肿瘤细胞(如抗原特异性及活化的CD8+ T细胞、记忆T细胞和克隆性T细胞)的直接作用可能引起肿瘤微环境的免疫启动。此外,在BRAFV600E黑色素瘤中联合BRAFi/MEKi和PD-1/PD-L1具有良好的疗效和持久的抗肿瘤反应。
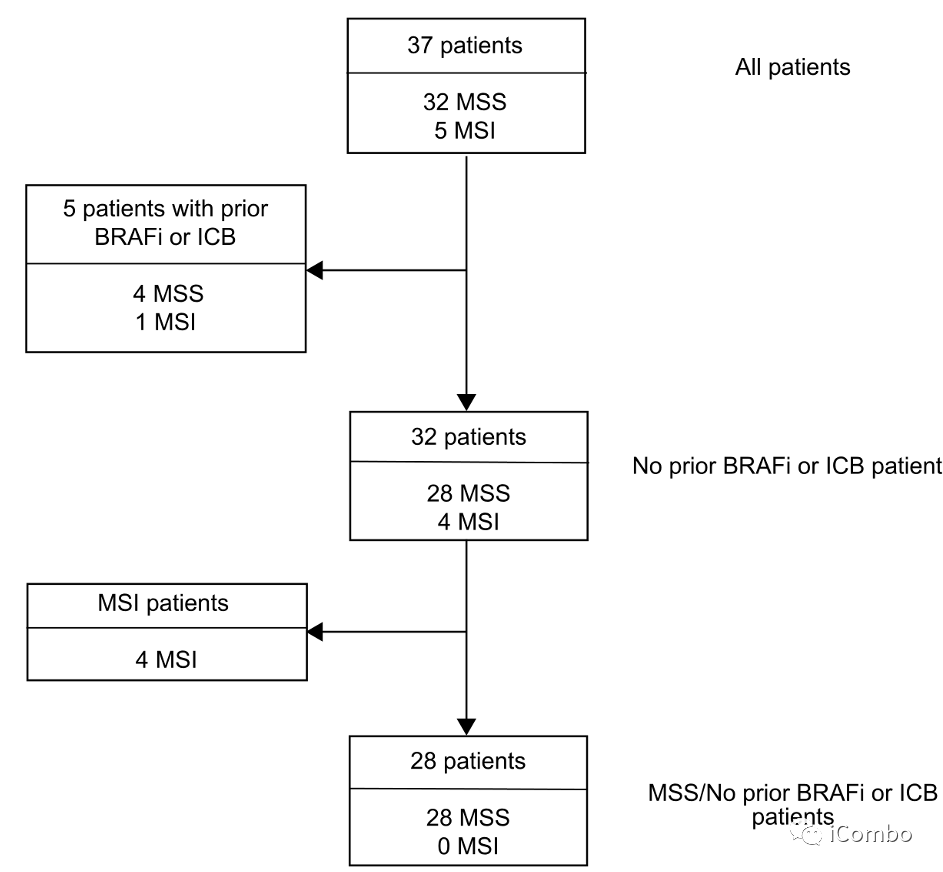
基于以上结果,该团队就MAPKi和免疫检查点抑制剂联合用药进行了II期临床试验(NCT03668431),BRAFi达拉菲尼联合MEKi曲美替尼和PD1抗体sparatlizumab(PDR001)治疗37例(32例为MSS 型)BRAF V600E 转移性CRC。ITT人群ORR为24.3%,达到预设主要研究终点。其次,DCR为70.3%,mPFS 为4.3个月,mOS为13.6个月。引人注目的是,在28例MSS型未经过靶向或免疫治疗的患者中,ORR为25%,DCR %,mPFS 为5 个月。
转化研究观察到,由于自适应反馈机制,单独的BRAF或MEK抑制无法维持CRC中MAPK通路的抑制作用。BRAF/MAPK抑制和免疫应答之间有潜在的协同机制,有效抑制MAPK通路以肿瘤细胞固有的方式驱动免疫基因表达的诱导,而不依赖于肿瘤免疫微环境的刺激。通过更优化的MAPK通路靶向组合来改善MAPK抑制,可以促进更大的免疫协同性,并与ICB联合获得更好的临床疗效(Nat Med.2023 Feb; 29(2): 458-466. doi: 10.1038/ s41591-022-02181-8)。
研究背景
BRAFV600E突变发生在约10%的结直肠癌(CRC)中,从而驱动MAPK信号通路的持续激活。携带BRAFV600E突变CRC患者预后不良,对标准治疗反应较差,中位总生存期(OS)仅为BRAF野生型患者的一半。
BRAF抑制剂,包括vemurafenib和dabrafenib,对BRAFV600E突变黑色素瘤非常有效,有效率可达60%-80%,而BRAFi单药治疗BRAFV600E CRC的有效率仅为0-5%。
既往研究指出,在BRAF被抑制后,强大的自适应反馈网络可以迅速重新活化MAPK信号通路。因此基于BRAFi联合多种靶向MAPK反馈通路药物的临床研究被设计进行,以期提高BRAFV600E结直肠癌患者的应答率。值得一提的是,BRAFi+EGFRi,BRAFi+MEKi,以及BRAFi+EGFRi+MEKi 这三种联合策略已有临床数据支持。近期FDA批准了BRAFi Encorafenib和抗EGFR抗体cetuximab联合用于治疗BRAFV600E晚期结直肠癌,然而该方案的客观缓解率仅为20%,中位无进展生存期(PFS)仅为4.3个月。因此,迫切需要探索新的有效治疗方法。
免疫检查点抑制剂在约4% MSI-H/dMMR的转移性结直肠癌中应答率约为40%,而在转移性 MSS CRC中的响应率几乎为0%。约15-20%的BRAFV600E转移性结直肠癌为MSI,这类患者对BRAF/MAPK通路抑制剂的反应比MSS患者更好且更持久,这些数据提示BRAF通路抑制可能会增强BRAFV600E CRC的免疫应答。
临床前研究表明,联合MAPK抑制和ICB可增强BRAF和KRAS突变型肿瘤的疗效,其潜在的协同作用机制包括BRAFi和/或MEKi对非肿瘤细胞(如抗原特异性及活化的CD8+ T细胞、记忆T细胞和克隆性T细胞)的直接作用可能引起肿瘤微环境的免疫启动。此外,在BRAFV600E黑色素瘤中,联合BRAFi/MEKi和PD-1/PD-L1具有良好的疗效和持久的抗肿瘤反应。
基于以上结果,该团队拟研究BRAFi/MEKi联合ICB在BRAFV600E CRC中的潜在协同作用。就MAPKi和免疫检查点抑制剂联合用药进行了II期临床试验,并对机制做了一定研究。
第一部分
MAPK inhibition enhances immune response in BRAFV600E CRC
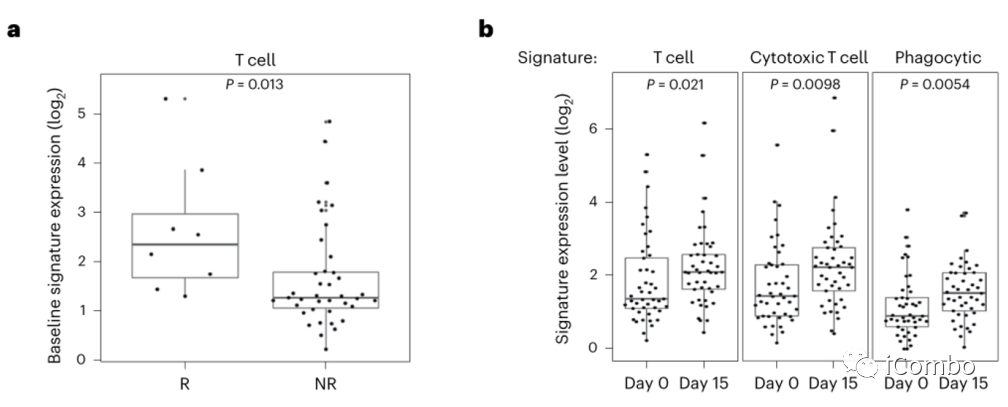
作者分析了BRAFV600E CRC患者早期联合BRAF/EGFRi±MEKi临床研究中71例患者的RNAseq数据,包括45例配对活检(基线和治疗第15天)和26例基线活检。
基线 RNAseq显示,相比无应答者,应答者的T细胞明显增多(图1a)。
在所有患者中,治疗15天后,相对于基线水平,T细胞、细胞毒性T细胞和其他免疫特征均有所增加,表明BRAF通路抑制后肿瘤中T细胞和免疫浸润增加(图1b) 。
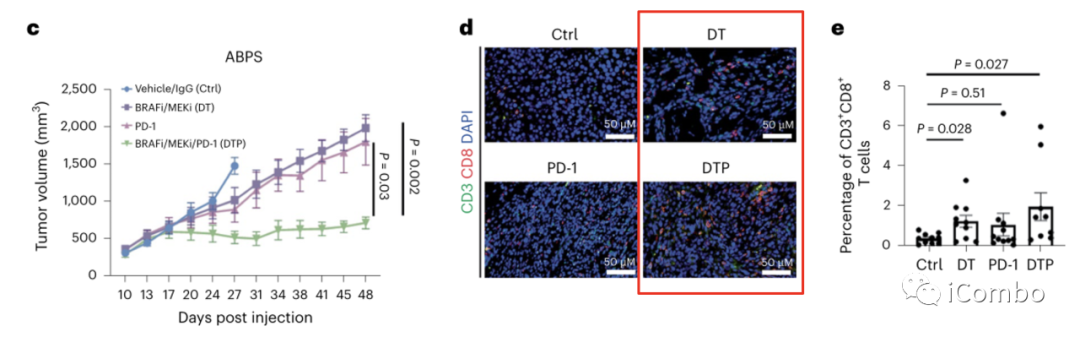
为了模拟这种潜在的协同作用,作者用C57BL/6结肠类器官,敲除APC、TP53和SMAD4,并表达BRAFV600E(ABPS细胞),构建了同源BRAFV600E CRC小鼠肿瘤模型。
小鼠移植瘤实验显示,联合使用BRAFi/MEKi/PD-1抑制剂可以更显著、更持续地减少肿瘤生长(图1c)。
在治疗10天后的肿瘤中,BRAF/MEKi处理导致CD3+CD8+ T细胞的百分比显著增加(图1d, e),单独使用PD-1抗体并没有导致CD3+CD8+ T细胞明显增加,但联合使用BRAFi/MEKi/PD-1抗体进一步使CD3+CD8+ T细胞显著增加(图1d, e)。
第二部分
Clinical efficacy
基于前期数据,作者启动了首个临床试验,探究BRAFi dabrafenib、MEKi trametinib和抗pd-1抗体spartalizumab(PDR001)治疗BRAFV600E CRC患者的疗效及安全性(概念验证II期单臂临床试验)。
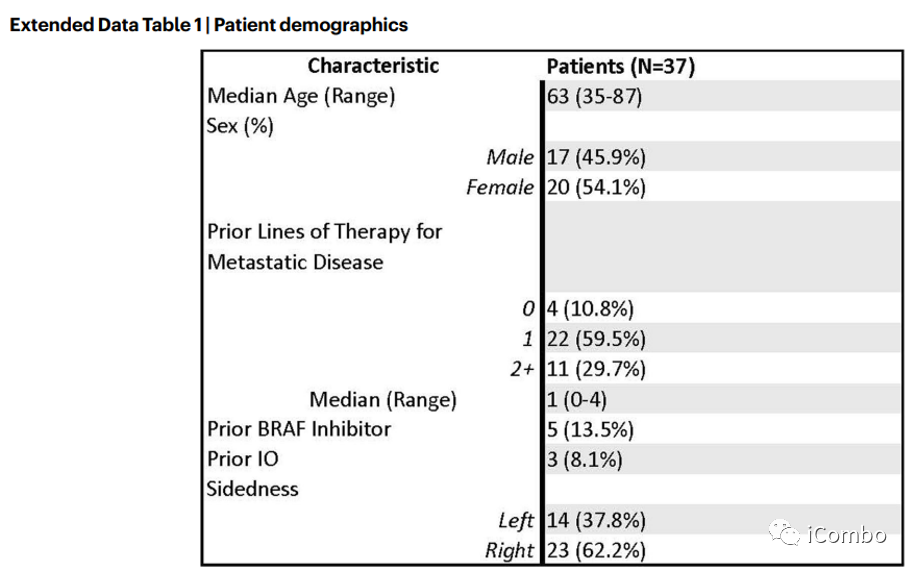
计划入组40例BRAFV600E CRC患者,至数据截止,已入组37例。5例患者既往接受过BRAFi和/或免疫检查点抑制剂治疗。中位随访时间为995天(245-1324)。主要研究终点 ORR,次要终点PFS、DCR、DOR和OS。
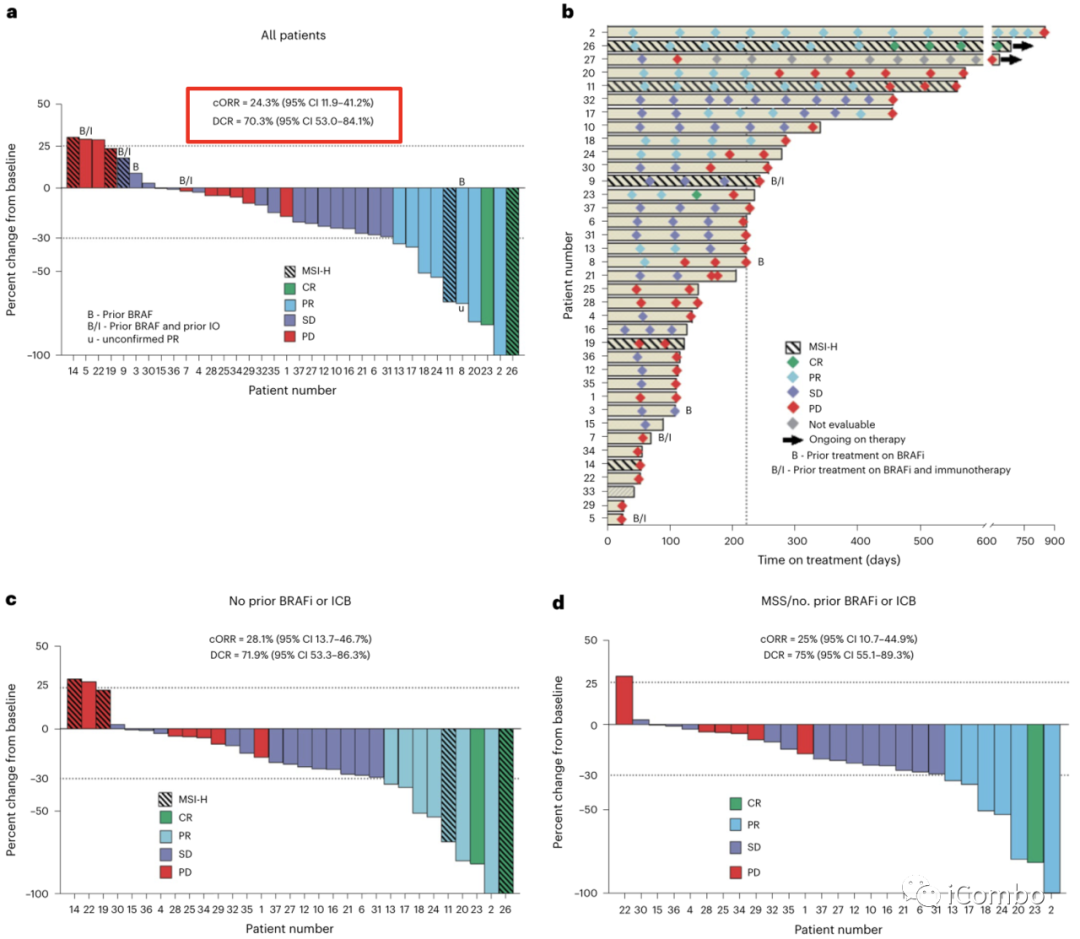
在37例患者中,9例获得了确定的缓解, 其中2例患者达到CR(ORR: 24.3%; 95%CI: 11.9-41.2%),另有1例患者获得未证实的缓解,DCR为70.3%(95% CI: 53-84.1%)(图2a),与BRAFV600E CRC中单独使用达拉非尼加曲美替尼的历史数据相比,有显著提高( cORR: 7% )。
中位治疗持续时间为7.4个月(95% CI: 4.2-7.9个月)(图2b)。
在32例既往未接受BRAFi或ICB治疗的患者中,cORR为28.1%,DCR为71.9%(图2c)。
在MSS BRAFV600E CRC患者中,cORR为25%(95% CI: 10.7-44.9%),DCR为75%(95% CI 55.1-89.3%)(图2d),再次优于历史对照。

中位PFS为4.3个月(95% CI 3.7-7.3个月),中位OS为13.6个月(95% CI 8.2-16.5个月)。
在MSS BRAFV600E 既往未接受过BRAFi或ICB治疗的CRC患者中,中位PFS为5个月(95% CI 3.7-7.4个月),5名患者(18%)持续接受治疗超过一年,1名患者获得了持续2.5年的PR。既往研究中单独使用DT的中位PFS仅为3.5个月,没有MSS患者持续治疗超过1年
左半和右半原发肿瘤没有观察到疗效的显著差异。
既往接受过BRAFi或ICB治疗的CRC患者的ORR为20%,PFS为3.7个月,OS为8.2个月。
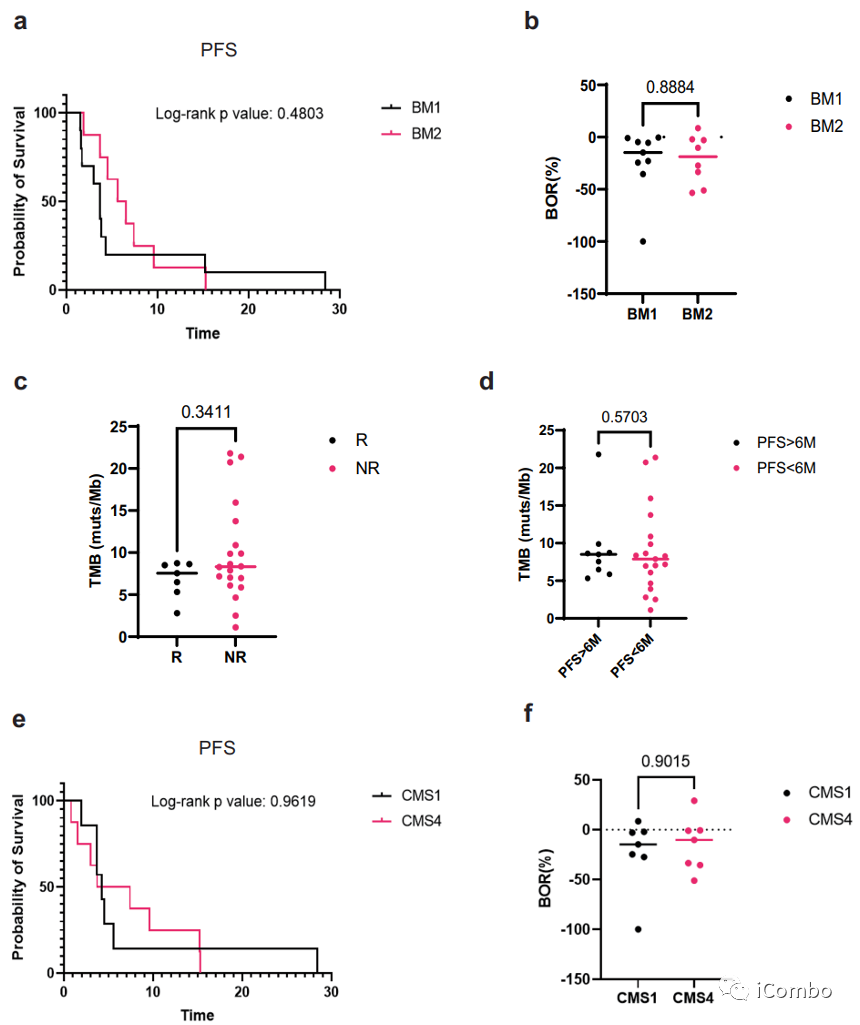
基线活检分析显示,之前被报道影响预后和治疗反应的因素,如TMB、BM1/BM2转录亚型和CMS分型,均与临床结果不相关。
BRAF转录亚型可以分为2类:BM1和BM2。BM1亚型与KRAS/AKT通路激活有关,因此对BRAF与MEK抑制剂敏感。BM2亚型细胞周期检查点下调,因此对CDK1抑制剂敏感。
第三部分
Tumor-intrinsic immune response and MAPK inhibition
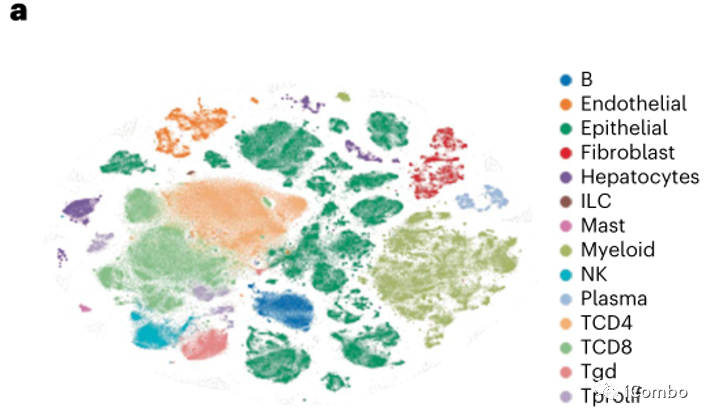
为了探究BRAF/MAPK通路抑制与肿瘤免疫反应的潜在相互作用,所有患者都进行了基线和治疗后第15天同一肿瘤的配对活检。用scRNAseq对23例患者的新鲜肿瘤组织进行分析,共有419,551个单细胞通过了质量控制(QC),这些细胞包括肿瘤、间质、免疫细胞和肿瘤上皮细胞群(图3a)。
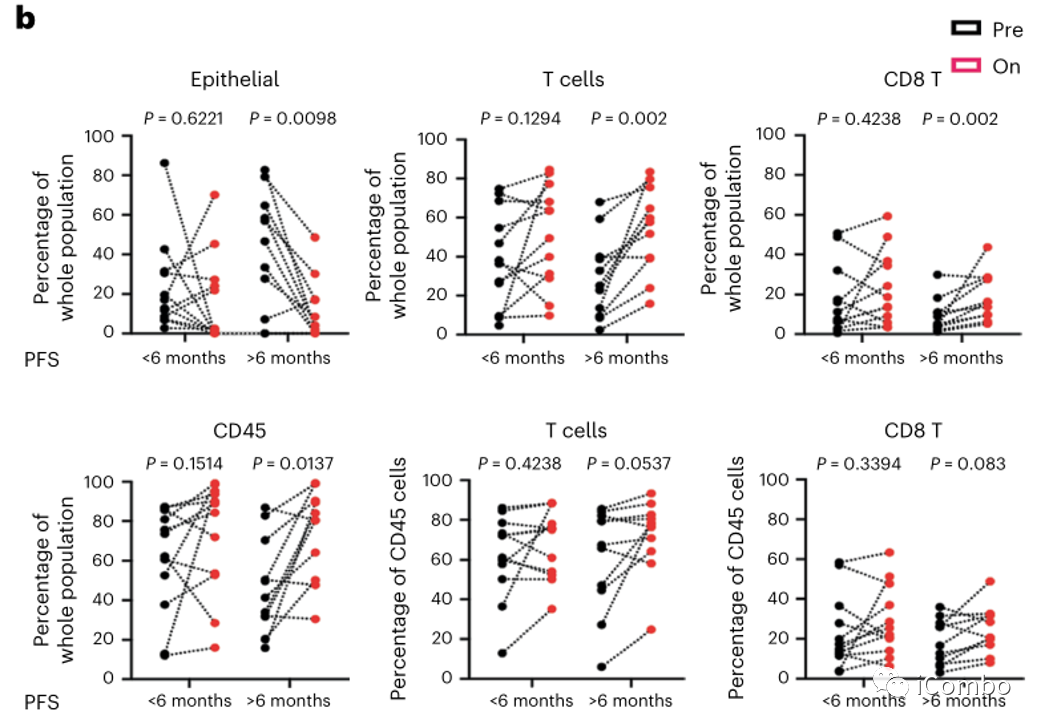
比较治疗前和治疗后单个细胞群丰度的变化,可观察到PFS > 6个月的患者(n=11)治疗后肿瘤上皮细胞显著减少,CD45+免疫细胞、T细胞和CD8+ T细胞显著增加,而PFS < 6个月的患者(n=12)治疗后细胞群无明显改变(图3b)。
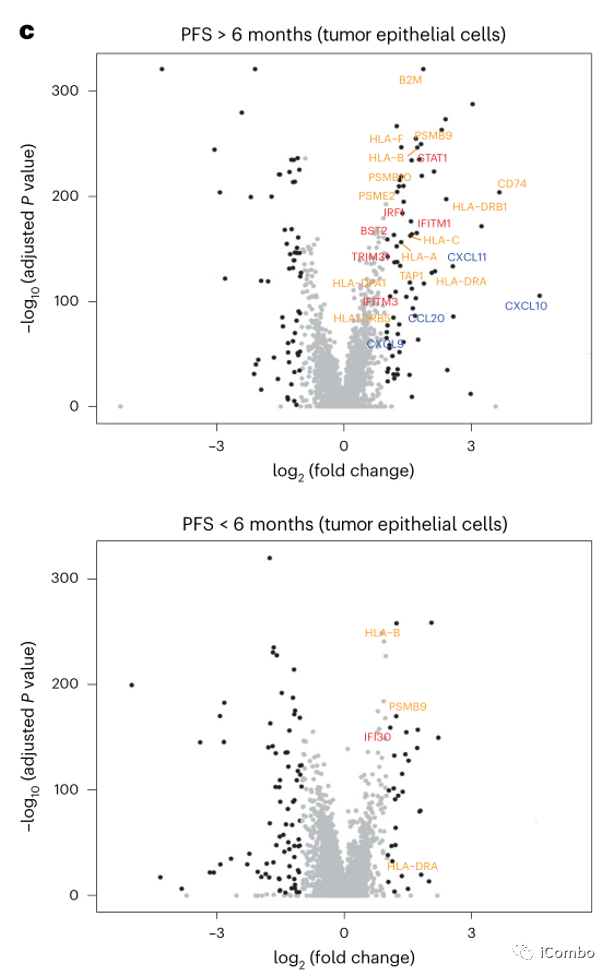
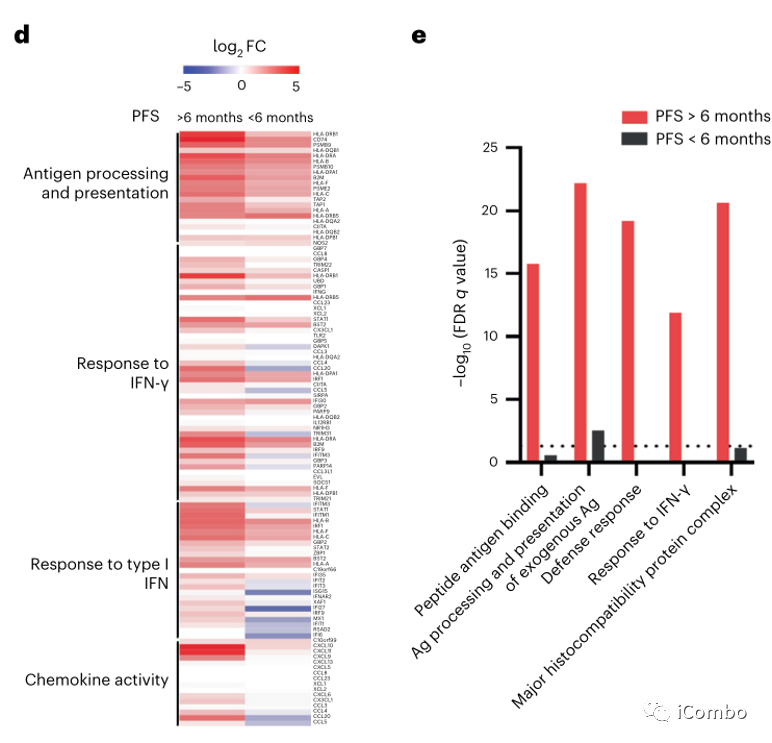
对比治疗前后差异表达基因,发现PFS > 6个月患者中免疫相关基因的表达显著增加,而PFS < 6个月患者中未观察到。这些基因包括参与干扰素(IFN)反应的基因(如STAT1, IRF1, IFITM1, IFITM3, BST2和TRIM31),抗原加工和递呈基因(如B2M, CD74, PSMB10, TAP1, HLA-A, HLA-C, HLA-F,HLA-DRB1和HLA-DPA1),趋化因子(例如CXCL9、CXCL10和CXCL11)(图3c,d),提示IFN刺激的转录程序和抗原加工和递呈途径的全面上调。
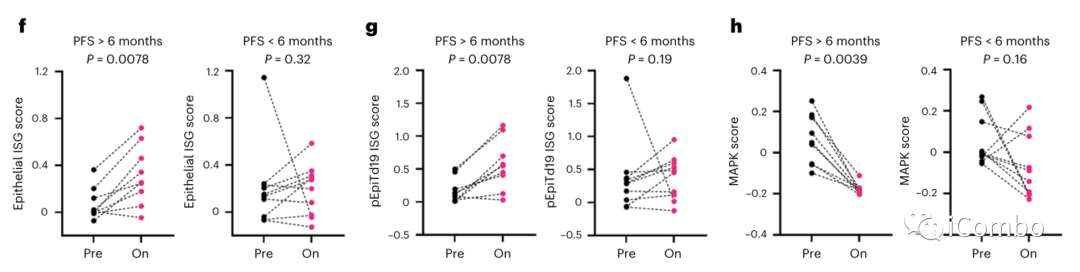
研究者进一步评估了人类结直肠癌恶性上皮特异性ISG程序的评分,该程序最近在一个独立的scRNAseq研究中获得,与I型干扰素反应和趋化因子活动相关,表征激活的T细胞。
PFS > 6个月的患者在治疗后第15天的评分(pEpiTd19 ISG)显著增加,而PFS < 6个月的患者则没有变化。
接下来利用MAPK基因表达签名分数检测了肿瘤上皮细胞中MAPK调节转录本(DUSP6, ETV4, ETV5和SPRY4)的变化,观察治疗前后肿瘤细胞中MAPK通路抑制情况。结果显示在PFS > 6个月患者的肿瘤上皮细胞中,MAPK评分在治疗后显著降低,而在PFS < 6个月的患者中则无明显变化(图3h)。
上述结果提示肿瘤固有免疫程序的诱导及MAPK通路的抑制与更长的PFS相关。
第四部分
Enhanced immune response driven by optimized MAPK inhibition
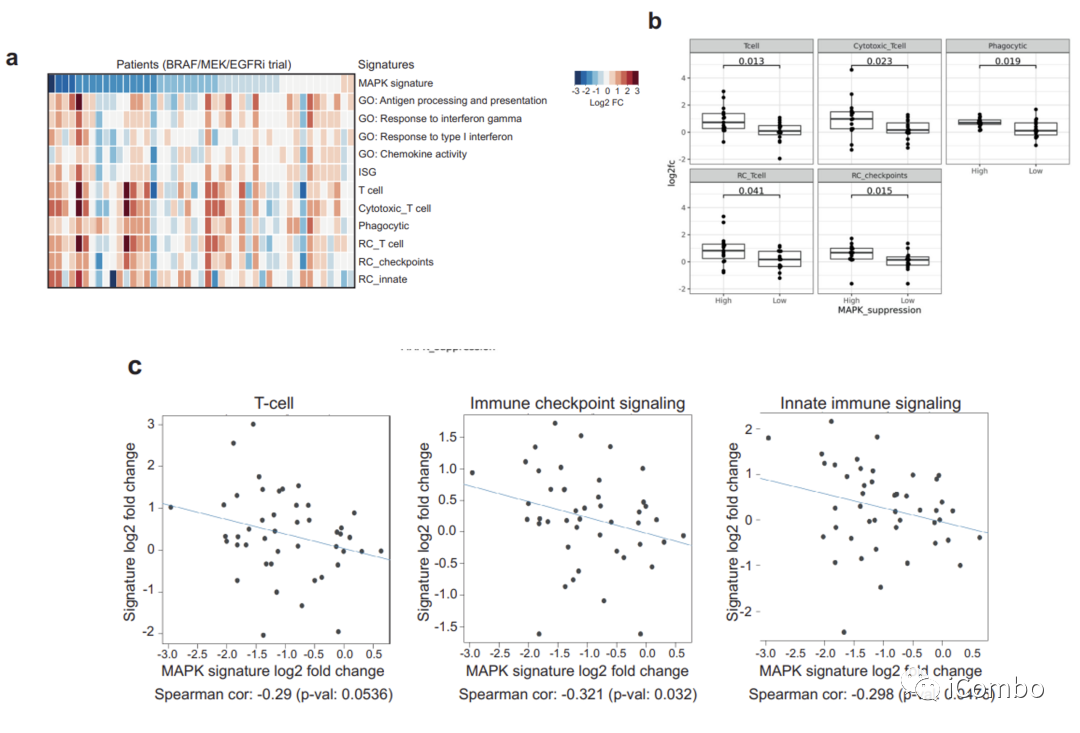
基于以上结果,假设在肿瘤细胞中MAPK抑制程度可能与肿瘤内在诱导免疫基因表达的程度直接相关。接下来分析了来自先前BRAF/MEK/EGFR抑制临床试验中BRAFV600E结直肠癌患者的45对基线和治疗后第15天活检组织的RNAseq数据。
结果显示,在治疗后第15天,免疫特征的诱导与MAPK通路抑制的程度相关(扩展数据图6a),这些信号包括T细胞和细胞毒信号、免疫检查点信号和吞噬相关信号(扩展数据图6b)。
此外,治疗前后MAPK抑制程度与免疫特征诱导之间存在相关性,包括T细胞、免疫检查点和先天免疫反应通路(扩展数据图6c)。在MAPK通路抑制程度较高的BRAFV600E CRC患者中观察到更强的免疫特征增加,表明肿瘤细胞中MAPK抑制程度可能驱动免疫基因诱导和肿瘤的免疫应答。
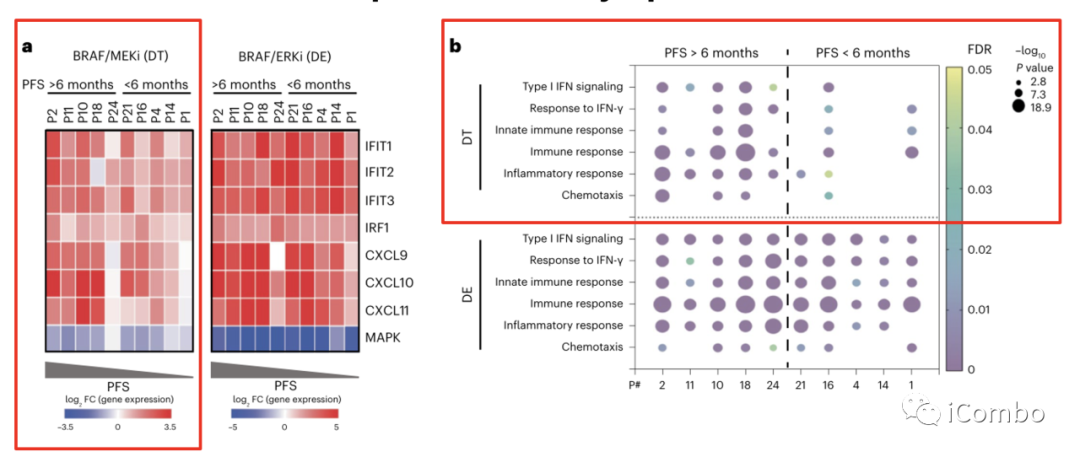
为了评估肿瘤细胞内MAPK抑制与免疫程序诱导之间的潜在关系,作者利用10例患者的基线肿瘤活检组织成功建立患者来源的类器官模型,其中5例PFS > 6个月,5例PFS < 6个月。
用DT处理类器官,qPCR检测基因表达。结果显示,与来自PFS < 6个月患者的类器官相比,PFS > 6个月患者的类器官在IFN反应相关基因(IFIT1, IFIT2, IFIT3和IRF1)的表达和趋化因子(CXCL9, CXCL10和CXCL11)活性方面有显著上调(图4a,左)。而PFS < 6个月患者的类器官在DT治疗后显示出较低程度的MAPK通路抑制(通过平均DUSP6, ETV4, ETV5和SPRY4 log2FC计算)(图4a,左)。提示MAPK通路抑制不足可能是免疫上调差异的潜在原因。
对类器官的转录组学分析( RNAseq)显示,与来自PFS < 6个月患者的类器官相比,来自PFS > 6个月患者的类器官显著诱导了更多的免疫基因集(图4b)。
这些数据证实,MAPK通路抑制以肿瘤细胞固有的方式驱动免疫基因表达的诱导,而不依赖于肿瘤免疫微环境的刺激,因为类器官培养物只含有肿瘤细胞,不包含来自肿瘤微环境的免疫细胞或基质细胞。

为了探究BRAF/MAPK通路的更强抑制是否可以增强所有肿瘤模型(包括PFS < 6个月的患者)的免疫基因诱导程度,作者进一步使用了BRAFi+ERKi的组合。
当用DE治疗时,所有类器官都表现出强大的MAPK抑制作用,无论来自类器官的患者的临床结局如何(图4a,右)。BRAF/ERK抑制在所有类器官中显著诱导了免疫应答基因的表达(图4a,右),这表明通过更完全的MAPK抑制可以实现更高的ISG诱导,特别是在对BRAF/MEKi反应较差的类器官中。
与BRAF/MEKi治疗相比,BRAF/ERK抑制剂(ERKi)处理普遍诱导PFS > 6个月和PFS < 6个月患者来源的类器官中免疫相关基因的强烈富集(图4b)。
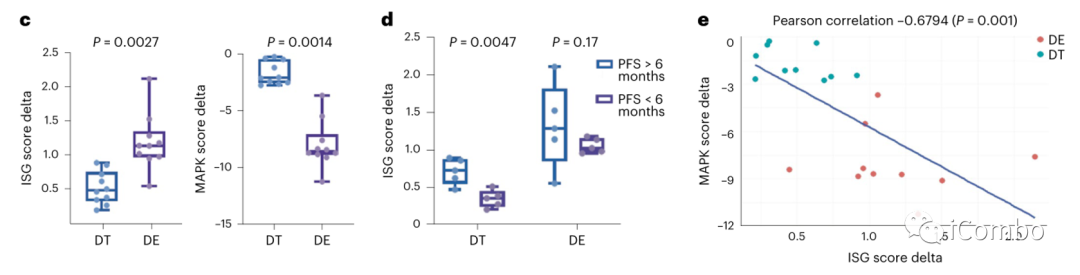
在所有类器官中,BRAF/ERKi诱导的ISG上调和MAPK抑制程度明显高于BRAF/MEKi(图4c)。
与PFS < 6个月的患者相比,BRAF/MEKi导致PFS > 6个月患者来源类器官的ISG评分显著增加,而BRAF/ERKi导致两组类器官的ISG评分诱导程度相当,无显著差异。两组患者在BRAF/ ERKi处理后的ISG评分诱导水平均等于或大于PFS > 6个月患者经BRAF/MEKi处理后的诱导程度(图4d)。
ISG评分与MAPK评分呈显著负相关(图4e)。
综上所述,这些结果表明:(1)单独有效抑制MAPK可诱导肿瘤细胞固有免疫基因表达;(2)提高MAPK抑制程度可使所有患者模型中肿瘤固有免疫基因表达水平达到与BRAF/MEKi处理后PFS > 6个月患者相同的水平。
第五部分
Toxicity AE summary
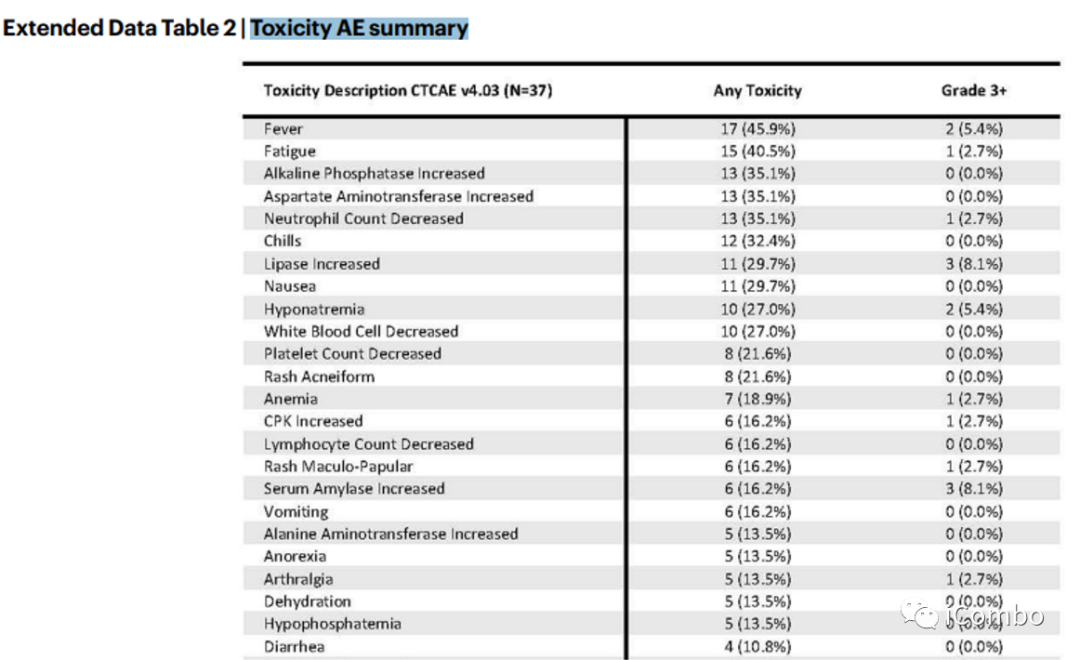
总体而言,该方案耐受性良好;3级以上毒副反应发生率较低;
皮疹、发烧、腹泻、疲乏、转氨酶升高是最常见的不良事件。
结论及研究意义
由于自适应反馈机制,单独的BRAF或MEK抑制无法维持CRC中MAPK通路的抑制作用;
BRAF/MAPK抑制和免疫应答之间有潜在的协同机制,有效抑制MAPK通路以肿瘤细胞固有的方式驱动免疫基因表达的诱导,而不依赖于肿瘤免疫微环境的刺激;
通过更优化的MAPK通路靶向组合来改善MAPK抑制,可以促进更大的免疫协同性,并与ICB联合获得更好的临床疗效。

