专家论坛|张继平:胆汁淤积性肝病的病理学诊断
时间:2024-07-05 16:01:51 热度:37.1℃ 作者:网络
胆汁淤积性肝病(CLD)依据部位分为肝内胆汁淤积和肝外胆汁淤积,肝内以免疫、遗传、药物/毒物性CLD为多见,肝外以梗阻、感染、创伤、肿瘤等为多见。依据病程分为急性和慢性CLD,急性CLD病程<6个月,以胆汁淤积为主,可见于药物性肝损伤(DILI)、急性感染、移植排异反应等;慢性CLD病程>6个月,以胆盐沉积为主,可见于各种慢性肝病及肝硬化。
1CLD常见的病理学特征
CLD常见病理学特征包括肝细胞及毛细胆管淤胆、胆管病变、胆盐沉积、肝细胞变性、胆汁性梗死及胆汁湖。淤积的胆汁因成分及氧化程度的不同,可呈淡黄色、黄褐色、墨绿色、棕红色、黑褐色等,淡黄色较稀薄的胆汁常见于急性胆汁淤积,墨绿色浓缩的胆汁可见于胆道梗阻。
1.1 肝细胞胆汁淤积
肝细胞内胆红素颗粒淤积,多分布在毛细胆管周围。不同病变形成的胆红素颗粒的形态特征常常不同,黄褐色细颗粒状无折光性脂褐素见于Gilbert综合征,黑褐色粗大颗粒状、van Gieson染色阴性见于Dubin-Johnson综合征,而黄褐色有折光性粗颗粒见于含铁血黄素,以腺泡1区为明显,向中央静脉逐渐递减,普鲁士蓝染色阳性,与以上不同。
1.2 毛细胆管淤胆
可见毛细胆管扩张,管腔内胆汁淤积,胆汁浓缩形成胆栓,常伴有肝细胞淤胆及Kupffer细胞活化,过碘酸雪夫染色(D-PAS染色)示较多蜡质样细胞。轻度胆汁淤积以肝腺泡3区为主,重度可累及全小叶。
1.3 胆管病变
胆管病变包括胆管损伤/缺失、炎症、淤胆。损伤的胆管上皮细胞大小不等,排列不整齐,胞质空泡或嗜酸化、萎缩、脱落或增生,甚至胆管减少或缺失。胆管缺失时常见CK7阳性肝细胞,提示肝细胞胆管化,可伴或不伴细胆管反应,细胆管反应不良提示预后和治疗效果不佳。损伤的胆管管壁及管周可有炎症细胞浸润,以淋巴细胞、浆细胞浸润为主可见于免疫性胆管炎,而梗阻或感染性胆管炎以中性粒细胞、嗜酸性粒细胞浸润为主。胆管内也可见胆汁淤积及胆栓,胆管基底膜可被破坏或纤维性增厚,Masson、D-PAS染色有助于观察。
1.4 胆管板畸形
由于小叶间胆管板不完全重塑,导致胚胎期胆管过多、扩张、胆管板残余内陷伴纤维化而引起胆管板畸形。典型表现为密集的小胆管排列在汇管区周边,汇管区中央为纤维血管及内陷的胆管板,扩张的胆管形态不规则,也可呈囊性扩张,管腔内淤胆及胆栓,胆管细胞体积较小,呈扁平状。
1.5 肝细胞变性
胆汁淤积时,可见肝细胞气球样及羽毛状变性,也可见巨细胞变肝细胞。气球样及羽毛状变性肝细胞明显肿胀,前者胞质淡染颗粒状,后者呈丝网状,可见淤胆性色素颗粒。巨细胞变肝细胞胞体巨大,胞质丰富,多核(图1),常见于婴幼儿,少见于成人,可能是未成熟的肝细胞对各种损伤(如胆汁淤积)的反应,其形成有2种可能的机制:(1)单个细胞相互融合而成合体细胞;(2)核分裂时细胞质未分裂。
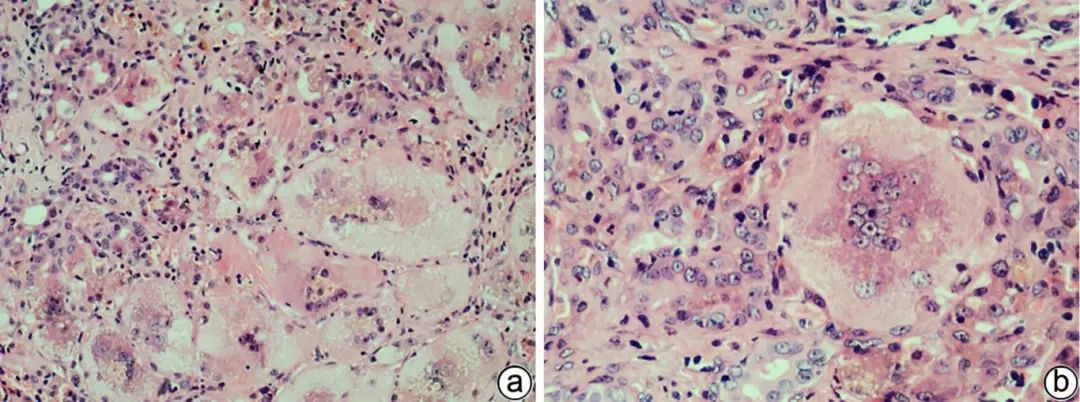
注:a,×100;b,×200。较多巨细胞变肝细胞,胞体巨大,多核,胞质内可见淤胆性色素颗粒,汇管区小胆管损伤及轻微扩张,间质可见中性粒细胞等炎性细胞浸润。
图1 巨细胞变肝细胞(HE染色)
1.6 胆汁性梗死及胆汁湖
胆汁淤积导致肝细胞梗死,梗死灶内肝细胞边界模糊不清,淡黄色素沉积,网状纤维塌陷或消失,胆汁外溢充满坏死区形成“胆汁湖”,伴炎症细胞浸润,巨噬细胞及多核巨细胞反应并吞噬色素。小的梗死灶可见于各种胆汁淤积,邻近汇管区大的梗死灶及胆汁湖高度提示大胆管梗阻。
1.7 胆盐沉积
腺泡1区肝细胞内细颗粒状胆盐沉积,可见气球样、羽毛状变性肝细胞,罗丹宁、地衣红染色可见铜颗粒沉积。
2CLD的病因及病理学诊断
CLD病因众多且复杂,可因同形不同,亦可形同因不同。依据病因,详细阐述CLD的病理学模式,有助于CLD的病因病理学诊断,最终诊断需要结合临床病史、实验室检测、影像学、基因检查等综合分析。
2.1 自身免疫性胆管炎
自身免疫性胆管炎为慢性CLD,包括:原发性胆汁性胆管炎(PBC)、原发性硬化性胆管炎(PSC)、IgG4相关硬化性胆管炎(IgG4-SC),均可表现胆管损伤及管周活化的T淋巴细胞浸润,可进展为肝硬化及其并发症,而每种疾病病理学又有相对特异性。
2.1.1 PBC
PBC多见于中老年女性,常伴ALP水平升高,病理主要为小叶间胆管损伤伴淋巴细胞、浆细胞包围浸润,旺炽型胆管炎(胆管上皮胞质嗜酸性化,核固缩)是PBC的独特病变(图2a、b),浸润的淋巴细胞以CD4+、CD8+ T淋巴细胞为主,随病情进展,小胆管被破坏消失,浆细胞在胆管周围呈冠状浸润是PBC的特征。多少不等的嗜酸性粒细胞浸润及细胆管反应常见于PBC早期,淋巴滤泡可见于早期和晚期。胆管上皮也可增生呈层状,胆管也可因基底膜被破坏而扩张。典型的PBC,损伤的胆管周围有时可见非干酪性上皮样细胞肉芽肿(图2c),需要与感染和结节病等鉴别,结合D-PAS、六氨银、吉姆莎、抗酸染色以及病原微生物宏基因检测、临床病史常可明确诊断。晚期PBC小胆管减少或缺失明显,伴纤维化或肝硬化,炎症病变轻微,诊断较困难,而观察到CK7阳性肝细胞及铜沉积,可提示PBC,但需除外PSC、DILI、Alagille综合征等,结合病史及胆管酶、AMA、M2、SP100、gP210、IgM检测可予以鉴别。
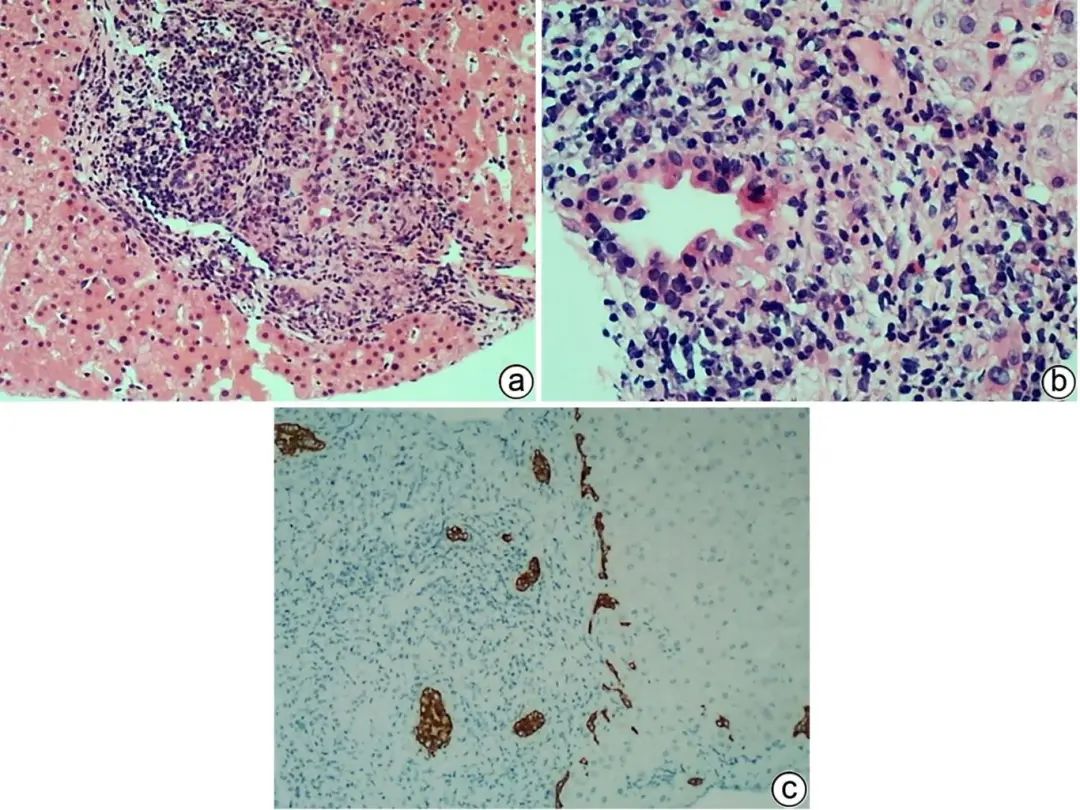
注:a,小胆管损伤伴淋巴细胞、浆细胞包围浸润及上皮样细胞肉芽肿,偶见多核巨细胞(HE染色,×100);b,旺炽型胆管炎,胆管上皮细胞嗜酸性化,核固缩,排列不整齐,管壁及管周较多淋巴细胞、浆细胞聚集浸润,散在少许嗜酸性粒细胞(HE染色,×200);c,CK7标记未见小胆管缺失,可见轻微细胆管反应(免疫组化,×100)。
图2 PBC典型的病理学特征
2.1.2 PSC
PSC可发生于各年龄段,以年轻男性最常见,与炎症性肠病密切相关,也与胃肠道恶性肿瘤(胆管癌、结直肠癌)发生风险有关。
PSC多发生于大胆管,胆管周围“洋葱皮样”纤维化伴淋巴细胞、浆细胞浸润,胆管细胞萎缩,管腔逐渐狭窄,甚至闭塞呈胆管样瘢痕,是PSC的病理特征;而小胆管的PSC表现为小叶间胆管基底膜增厚伴透明变性(图3),即小胆管硬化。部分PSC可见小动脉增大及管壁纤维性增厚。少部分PSC与PBC病变有重叠,肝组织学鉴别比较困难,然而,与PBC相比,PSC胆汁淤积更为频繁、出现更早,管周纤维化、胆管瘢痕,常常提示PSC,而肉芽肿最常见于PBC。需要注意的是,“洋葱皮样”纤维化及小胆管硬化亦可见于继发性硬化性胆管炎。
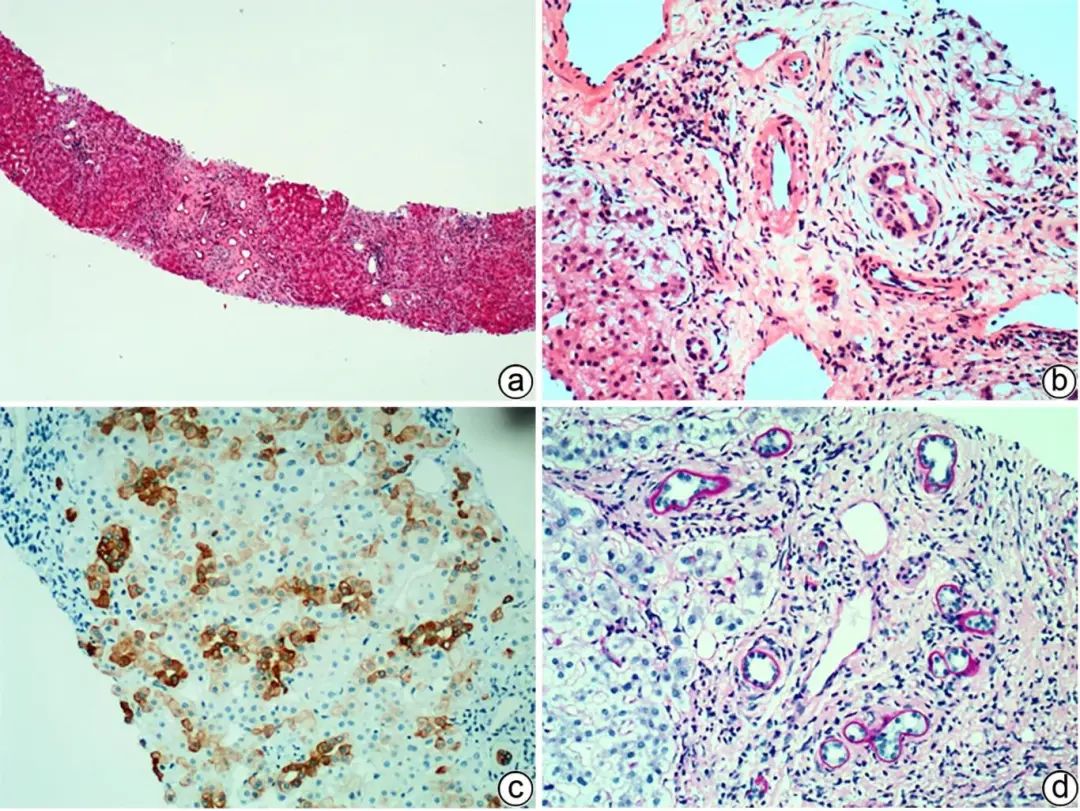
注:a,肝硬化,汇管区轻度炎症,小胆管轻度扩张,管周纤维化(HE染色,×20);b,汇管区轻度炎症,小胆管基底膜增厚,周围“洋葱皮样”纤维化,小动脉扩大,管壁轻微增厚,门静脉分支扩张及管周纤维化,汇管区周围肝细胞气球样变性(HE染色,×100);c,免疫组化CK7显示肝细胞胆管化(IHC染色,×100);d,小胆管基底膜增厚(D-PAS染色,×100)。
图3 PSC典型的病理学特征
2.1.3 IgG4-SC
IgG4-SC是一种对类固醇治疗有效、临床预后良好的硬化性胆管炎,多发于中老年人,常并发自身免疫性胰腺炎、腮腺炎等多器官病变,血清IgG4水平升高,也可见CA19-9升高,影像学检查示胆管壁增厚、管腔狭窄或占位病变样,需要与PSC、胆管癌、继发性硬化性胆管炎鉴别。IgG4-SC典型病理特征:胆管炎伴较多IgG4+浆细胞和淋巴细胞浸润,席纹状纤维化,闭塞性静脉炎。手术切取肝组织IgG4+浆细胞>50个/HPF,穿刺活检IgG4+细胞>10个/HPF,IgG4/IgG>40%是IgG4-SC的诊断标准之一。然而,由于IgG4-SC常发生于大胆管,肝穿刺活检席纹状纤维化、闭塞性静脉炎常不典型或缺乏,需结合血清IgG4水平、有无多器官病变及类固醇治疗应答情况等综合考虑。
2.2 遗传代谢性肝病
遗传代谢性肝病胆汁淤积是由于基因突变及编码蛋白异常而导致的,包括:遗传性非溶血性高胆红素血症、胆汁酸代谢异常、铜和铁代谢紊乱、氨基酸代谢异常等,多有特异性。
2.2.1 遗传性非溶血性高胆红素血症
遗传性非溶血性高胆红素血症是肝细胞性CLD,临床表现为反复黄疸,多数无远期进展,包括:Gilbert综合征(GS)、Crigler-Najjar综合征(CNS)、Dubin-Johnson综合征(DJS)、Rotor综合征(RD)。GS和CNS是肝内UGT1A1活性降低或完全缺失引起的,以间接胆红素水平升高为主。DJS与RD以直接胆红素水平升高为主。
GS病理可见腺泡3区肝细胞内较多脂褐素样色素颗粒,分布在毛细胆管周围,可有少许铁颗粒沉积。透射电镜(TEM)可见色素颗粒透亮区大于高电子致密区,毛细胆管轻度扩张,尽管以上表现不具特异性,但对于青少年反复发作的不明原因黄疸,以间接胆红素水平升高为主,需要考虑GS的可能,苯巴比妥反应、UGT1A1基因突变可予以明确诊断。
CNS分为Ⅰ和Ⅱ型,CNSⅠ型UGT1A1完全缺失,病情最严重,可发展为核黄疸,甚至危及生命。病理学表现为肝内较重的胆汁淤积及胆栓,胆管损伤及细胆管反应,不同程度的炎症及纤维化。
DJS肝组织外观呈黑色,光镜下肝细胞内较多粗大的黑褐色淤胆性色素颗粒,呈带状分布在扩张的毛细胆管周围,TEM示色素颗粒高电子致密区大于透亮区,可检测到ABCC2基因突变。
RD患者肝细胞内色素颗粒极少,不易识别,TEM可见肝细胞胞质内高尔基体明显增生、肥大,结合SLCO1B1和SLCO1B3基因测序可明确诊断。
2.2.2 进行性家族性肝内胆汁淤积症(PFIC)
PFIC是一组常染色体隐性遗传病,多于婴幼儿或儿童期起病,亦可见于部分成人患者。常见临床表现有黄疸、瘙痒、脾大、门静脉高压、腹水,甚至肝硬化、肝衰竭。PFIC分为1~6型,分别由ATP8B1/PIC1、ABCB11/BSEE、ABCB4/MDR3、TJP21ZO2、NR1H4/FXR及MYO5B/BSEP基因突变及编码蛋白缺陷导致。
PFIC3主要表现为高GGT水平胆汁淤积、胆石症、肝纤维化。病理学表现主要为胆管的损伤、减少/缺失及增生,TEM可见胆固醇结晶。胆管缺失的程度与疾病严重程度和预后有关,胆管缺失比例越多,预后越差,炎症及纤维化越重。免疫组化MDR3显示毛细胆管减少或缺失,有助于诊断,但正常表达也不能除外PFIC3。ABCB4基因突变还可以引起妊娠期肝内胆汁淤积症、低磷脂相关性胆石症,严重程度低于PFIC3型。
除PFIC3外,其余5型均表现为低GGT水平胆汁淤积。病理表现肝细胞及毛细胆管明显淤胆及胆栓,不同程度炎症及纤维化,可有小胆管损伤及肝细胞胆管化,小胆管增生常不明显。PFIC1免疫组化毛细胆管膜上的FIC1蛋白缺失,TEM可见粗颗粒状胆汁,又称“Byler bile”。PFIC2主要为巨细胞肝炎,肝细胞排列紊乱,免疫组化BSEP缺失,TEM可见毛细胆管内非晶体和丝状胆汁淤积,管腔面微绒毛消失。PFIC4可见少许巨细胞变及气球样变性肝细胞,免疫组化TJP2缺失,TEM可见毛细胆管紧密连接延长,缺少密集的闭锁小带。PFIC5可见巨细胞肝炎、胆汁淤积、胆管缺失及增生,免疫组化FXR及BSEP缺失,TEM无BSEP分布异常。PFIC6与PFIC2相似,免疫组化MYO5B可见粗大的颗粒,BSEP减少或缺失,TEM无BSEP分布异常。
良性复发性肝内胆汁淤积症(BRIC)是反复发作的严重黄疸和瘙痒,GGT水平轻度升高或正常,间期无任何症状。根据突变基因不同分为BRIC1型和2型,BRIC1型由ATP8B1基因突变影响FIC1蛋白所致,BRIC2型由ABCB11基因突变引起BSEP蛋白异常而导致。病理表现毛细胆管温和的胆汁淤积,炎症及纤维化轻微。
2.2.3 铜和铁代谢异常性肝病
肝豆状核变性(WD)是由于ATP7B基因突变引起铜代谢异常,过多的铜沉积在肝脏、神经、眼睛、肾脏等器官,致器官损害。临床表现多样:无症状肝病、肝硬化、肝衰竭、神经精神症状等,铜蓝蛋白低、血清游离铜及24 h尿铜高、眼睛K-F环对诊断很重要,但在WD中并不总是异常的,也有可能与非WD疾病重叠。WD肝病最常见,病理学表现有异质性,可表现各种肝病的模式:轻度非特异炎症、脂肪肝/脂肪性肝炎、病毒性肝炎样、自身免疫性肝炎样、急慢性重度肝炎伴随亚大块状/块状坏死、铜颗粒沉积等,鉴别诊断范围广,而汇管区周围糖原核肝细胞、气球样及嗜酸性颗粒状肝细胞、Mallory小体在WD更常见,TEM可观察到较弥漫的肝细胞内线粒体丰富、形态不规则、内外膜分离、内嵴扩张空泡及高电子致密颗粒(图4),常提示WD,而局灶线粒体病变也可见于PBC、线粒体病等,ATP7B基因检测非常有帮助,然而,约5%的WD可能检测不到可识别基因突变,因此,WD的诊断需要结合病史、肝组织铜含量测定、病理学检查、基因检查综合分析。
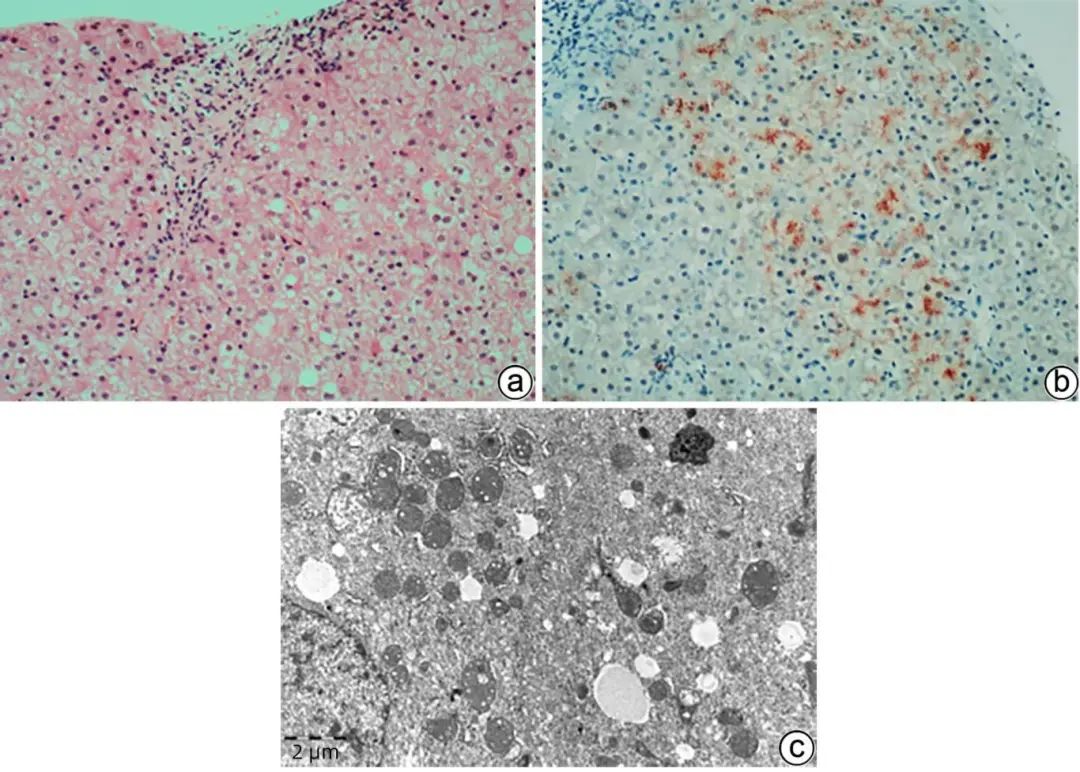
注:a,肝细胞水肿、脂肪变性、气球样变性,部分胞质呈淡红色颗粒状,腺泡1区少许糖原核肝细胞。汇管区轻度炎症(HE染色,×100);b,肝细胞胞质内较多红色铜颗粒沉积(罗丹宁铜染色,×100);c,肝细胞胞质内线粒体形态不规则,内外膜分类,可见内嵴扩张空泡及圆形高电子致密颗粒。肝细胞胞质内也可见脂滴、含高电子致密度的淤胆性色素颗粒沉积(TEM,×12 000)。
图4 肝豆状核变性的组织病理学及超微结构特征
血色病是因铁过载沉积于肝脏、胰腺、皮肤等,导致相应组织器官的损伤,包括原发性(遗传性)与继发性血色病,多发生于中老年男性,主要表现为肝功能异常、肝硬化、糖尿病、皮肤色素沉着等。原发性血色病依据基因突变位点及表型不同,分为4种类型:HFE(Ⅰ型)、HJV(ⅡA型)、HAMP(ⅡB型)、TFR2(Ⅲ型)、SLC40A1(Ⅳ型),前3型为常染色体隐性遗传病,Ⅳ型为显性遗传。西方白种人群主要是HFE中p.C282Y纯合突变引起,而中国人群少见此突变。病理学表现为肝细胞、胆管细胞、内皮细胞、巨噬细胞及间质细胞内可见棕黄色含铁色素颗粒沉积,普鲁士蓝染色阳性,肝细胞内铁沉积从汇管区至中央静脉逐渐递减,可见铁死亡,不同程度炎症及纤维化。与继发性血色病相比,原发性血色病更易出现胆管上皮内铁沉积,ALT水平显著升高,白细胞及血小板水平显著减少,血清铁水平及转铁蛋白饱和度显著升高。
2.2.4 Citrin缺陷导致的新生儿肝内胆汁淤积症(NICCD)
NICCD是SLC25A13基因突变引起的婴儿胆汁淤积、肝酶升高(AST>ALT)、高氨和高瓜氨酸血症、脂肪肝、满月脸等,黄疸通常在1岁时得到改善。病理学表现可见较弥漫的肝细胞脂肪变性、胆汁淤积、轻度铁沉积,虽然单个病变无特异性,但组合发生在婴儿时应考虑NICCD的可能,TEM可见肝细胞线粒体大小不等,形态不规则,内嵴扩张。经过高蛋白、高脂、低糖饮食管理的患儿,可仅存在脂肪肝,胆汁淤积不明显,与Citrin缺陷Ⅱ型相似。仅有少数无脂肪肝的个例报道,诊断需要结合病史、血氨基酸、尿有机酸及基因检测综合考虑。
2.2.5 先天性纤维囊性肝病
先天性纤维囊性肝病是一组以胆道纤维囊性改变为特征的遗传性疾病,主要由PKHD1基因突变引起。包括先天性肝纤维化(CHF)、Caroli病、胆管错构瘤,均可见胆管板畸形。CHF多见于青少年,常表现为门静脉高压、胆管酶升高、脾大、肝硬化、肝肾囊肿等,临床上分为门静脉高压型、胆管炎型、混合型、无症状型。病理以胆管板畸形及囊性扩张、“地图样”纤维化、门静脉狭窄或血管纤维化为典型病变(图5);不典型者畸形胆管板较少,小胆管扩张及血管纤维化不明显,与其他原因引起的胆管梗阻、淤胆不易鉴别。Caroli病是较大的段胆管和隔胆管的囊性扩张,合并CHF时称为Caroli综合征,更末端呈微瘤样密集排列的小胆管称为胆管错构瘤。
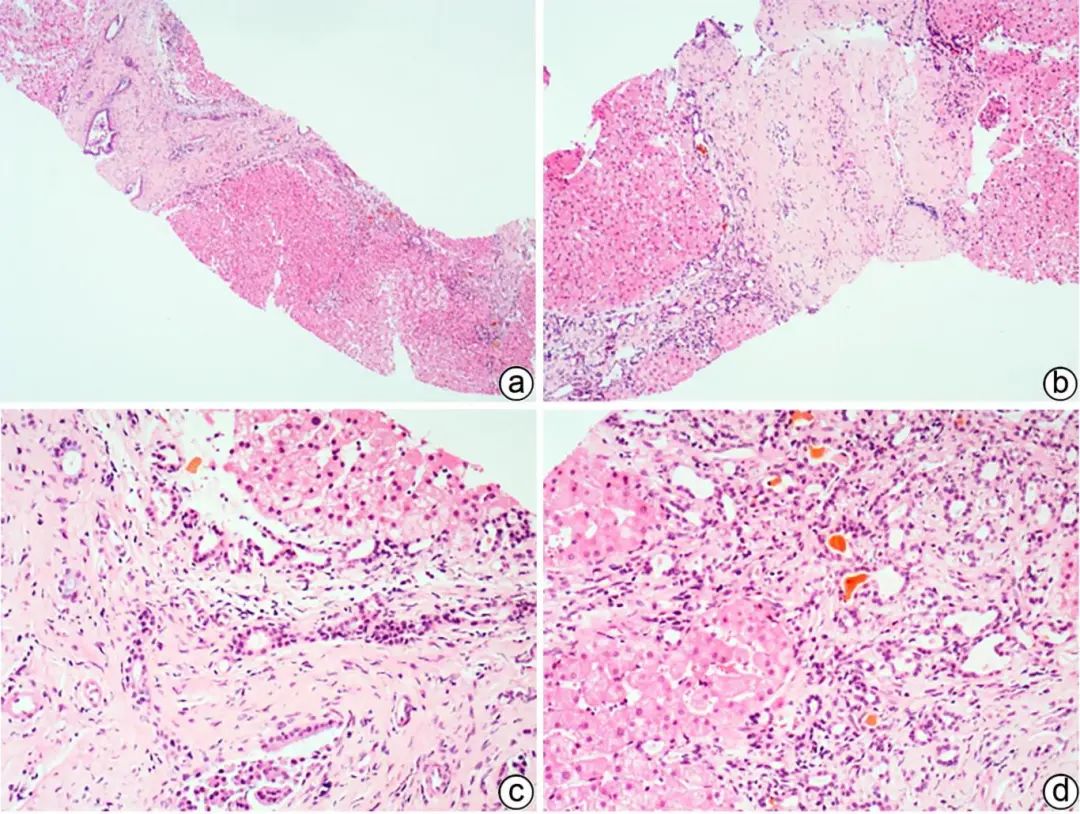
注:a,CHF“地图样”纤维化,胆管板畸形,胆汁淤积(HE染色,×40);b,汇管区胆管板畸形,门静脉狭窄、闭塞,肝细胞轻度水肿(HE染色,×100);c,畸形的胆管板内陷于汇管区纤维组织中,门静脉分支狭窄、闭塞(HE染色,×200);d,胆管板畸形,形态不规则,管腔扩张,可见胆栓,间质少许淋巴细胞、浆细胞浸润(HE染色,×200)。
图5 CHF典型的组织病理学特征
2.2.6 Alagille综合征
Alagille综合征是一种常染色体显性遗传病,与NOTCH信号通路受损有关,超过90%的患者由JAG1致病变异引起,少数由NOTCH2引起。肝脏受累最常见,以胆汁淤积、GGT及TBA水平升高为特征,伴脾大、生长发育迟缓、特殊面容(凸额、凹眼、鞍形鼻、颏小而尖、五官距离较宽)、心血管受累、蝶形椎骨、肾脏受累、角膜后胚胎环等多器官病变,部分患者有门静脉高压的临床表现。病理学特征:以胆管缺失及胆汁淤积、胆盐沉积为主,少部分患者有细胆管反应,可见CK7阳性肝细胞及铜沉积,不同程度的炎症及纤维化。胆管缺失及纤维化随年龄的增长而显著增加,目前少数发生肝癌的报道。本病需要与DILI、PBC等引起的胆管缺失综合征鉴别。
2.2.7 红细胞生成性原卟啉病(EPP)
EPP是罕见的遗传病,由血红素合成途径中亚铁螯合酶活性降低或缺乏,导致原卟啉Ⅸ在皮肤、肝脏等组织中过度积累而致病。主要临床表现为光敏性皮疹、黄疸、间歇性腹痛、神经肌肉病变、轻度贫血、维生素D缺乏等。EPP肝病更罕见,多为成年发病,表现为腹痛、肝功能异常、黄疸、胆石症等非特异症状,严重者出现肝硬化、肝衰竭。病理学表现除肝细胞水肿、气球样变性、脂肪变性、不同程度炎症及纤维化等非特异病变外,2种颜色的淤胆颗粒或胆栓具有特异性,其中一种为淡黄胆色素颗粒,另一种呈“巧克力”色,偏振光显微镜下呈红色双折光,中央可见“Maltese十字”或星状暗区(图6a、b),TEM淤积的胆汁中可见丝状弧形排列的原卟啉结晶(图6c、d),为EPP肝病较特征性改变。
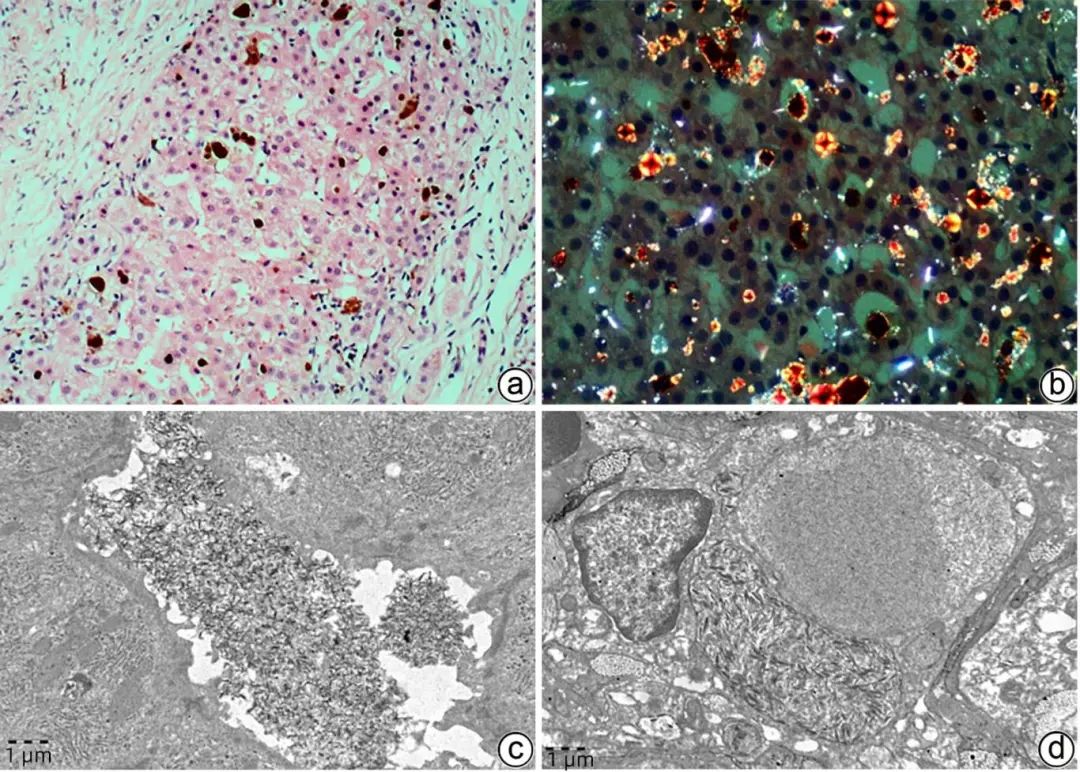
注:a,肝细胞及毛细胆管内两种颜色的淤胆颗粒及胆栓,一种“巧克力”色,另一种淡黄色(HE染色,×100);b,偏振光显微镜可见红色双折光中的“Maltese十字”或星状暗区(HE染色,偏振光,×200);c,毛细胆管内可见原卟啉结晶(箭头),呈丝状弧形排列,毛细胆管腔面微绒毛减少,紧密连接延长(TEM,×15 000);d,Kupffer细胞胞质溶酶体内充满丝状原卟啉结晶(箭头),窦周间隙可见胶原纤维束沉积(TEM,×15 000)。
图6 EPP的组织病理学及超微结构特征
2.3 大胆管梗阻性CLD
大胆管CLD主要以急性或慢性胆汁淤积为主,ALP、GGT水平常明显升高,总胆红素及直接胆红素水平升高为主。肝活检可见汇管区扩大,间质水肿,小胆管扩张、淤胆,可见胆栓,常伴小胆管增生及细胆管反应,腺泡1区肝细胞胆红素及胆盐沉积,可见气球样、羽毛状变性肝细胞,毛细胆管淤胆。淤胆的小胆管周围中性粒细胞、嗜酸性粒细胞浸润多见于大胆管梗阻及感染性疾病,如结石、寄生虫、胆汁螺杆菌感染、Oddi括约肌功能障碍等。细胆管胆汁淤积伴中性粒细胞浸润可见于脓毒血症。
胆道闭锁是肝内、外胆道系统进行性纤维化梗阻性胆管病,是新生儿和婴儿胆汁淤积常见的原因,患儿表现进行性黄疸、陶土色大便、小胆囊或胆囊缺如等,肝活检与其他大胆管梗阻病变相似,而小胆管、小动脉及门静脉周围较厚的纤维鞘、小胆管内致密的胆栓及汇管区边缘吻合的小胆管是有用的诊断特征。肝小叶内常见巨细胞变、腺泡状或淤胆性花结样肝细胞及髓外造血。汇管区周围少许肝细胞内可见铜沉积,少于50%的患儿可见胆管板畸形,少数可见胆管缺失。
2.4 其他CLD病变
2.4.1 DILI
DILI分为急性和慢性,急性DILI表现为肝炎、胆汁淤积及混合病变,诊断有挑战性,较明确的药物史或毒物接触史是重要的提示。急性淤胆型DILI以肝小叶内胆汁淤积为主,尤其是腺泡3区为明显,多见活化的Kupffer细胞及D-PAS染色阳性的蜡质样细胞,可见炎性坏死、嗜酸性变肝细胞,汇管区炎症及纤维化轻微,可见小胆管损伤。淤胆较重者可见胆汁性梗死、胆汁湖、胆管缺失。婴幼儿常见巨细胞变肝细胞。慢性DILI可表现为慢性肝炎、脂肪变性、磷脂质病、轻微胆汁淤积等,DILI的诊断需要排除病毒性肝炎、脂肪肝/脂肪性肝炎、PFIC等。
2.4.2 淋巴造血系统疾病或肿瘤
淋巴造血系统疾病或肿瘤如肝脾T细胞淋巴瘤、骨髓纤维化症引起的髓外造血,骨髓异常增生综合征、海蓝细胞组织细胞增生症、白血病或淋巴瘤累及肝脏,肝窦内较多有核血细胞或肿瘤细胞,肝淀粉样变性中淀粉样物质沉积在肝细胞间隙及窦周间隙,由于肝窦内压力增大,窦周间隙异常物质沉积,肝细胞被破坏等,均可引起胆汁的分泌和排泄受阻,导致肝细胞性CLD及门静脉高压,形态学特异,易于诊断。
3小结与展望
肝活检病理学检查在CLD的诊断中非常重要,可以发现潜在的病因或多因素疾病,评估胆汁淤积、炎症及纤维化的程度等。但目前肝活检存在一定局限性,且可用于诊断的特异性生物标志物较少。随着人工智能在医疗领域较广泛的应用,临床、病理、影像、基因、质谱、生化等多空间组学模态的研究将为CLD的精准诊疗带来光明的前景。
全文下载
https://www.lcgdbzz.org/cn/article/doi/10.12449/JCH240605


