成人II型瓜氨酸血症|病例分享
时间:2024-08-29 18:05:52 热度:37.1℃ 作者:网络
论坛导读:瓜氨酸血症(Citrullinemia)是一种罕见的遗传性代谢疾病,主要分为Ⅰ型和Ⅱ型。其中Ⅰ型瓜氨酸血症主要由于精氨琥珀酸合成酶(ASS1)基因突变导致,Ⅱ型则与谷氨酸氨基转移酶2(SLC25A13)基因突变相关。最新研究还发现了一些新的调控基因和代谢通路,它们在瓜氨酸血症的发病机制中可能扮演着重要角色。这种疾病通常由于尿素循环中的关键酶缺乏或功能异常,导致体内瓜氨酸及其他代谢产物的累积,影响氨的排出,进而引发一系列神经系统和肝脏症状。目前,瓜氨酸血症的治疗主要包括饮食管理和药物治疗。最新研究表明,个体化的氨基酸配方饮食和氨排出药物(如苯甲酸钠、苯丁酸钠)能够显著改善患者的预后。此外,基因治疗和肝脏移植也在研究中,未来有望为瓜氨酸血症患者提供更有效的治疗选择。
瓜氨酸血症的临床表现多样,Ⅰ型患者常在新生儿期发病,表现为厌食、呕吐、嗜睡和惊厥。Ⅱ型患者多在成年后发病,症状较轻,常表现为反复发作的肝功能异常和神经系统症状。对于瓜氨酸血症,早期诊断和个体化治疗至关重要。新生儿筛查能够及早发现高危患儿,并及时进行干预。此外,定期随访和监测可以及时发现疾病进展和并发症,确保患者获得最佳治疗效果。对于有瓜氨酸血症家族史的高危人群,应进行遗传咨询和产前诊断。由此可见,瓜氨酸血症是一种可防可治的罕见病,随着诊断技术和治疗手段的不断进步,患者的预后将得到显著改善。通过最新的指南和专家共识,医生和患者能够更好地了解和管理这一疾病,从而提高生活质量。
成人II型瓜氨酸血症(adult-onset type II citrullinemia,CTLN2)是由SLC25A13基因突变导致的神经遗传代谢病,为常染色体隐性遗传病。CTLN2最早由日本学者报道,在东亚地区更多见。该基因位于染色体7q21.3,日本、中国、泰国等亚洲国家最常见的突变类型为c.851-854del突变,其次为c.IVS11+1G>A、c.IVS6+5G>A、c.S225X等突变。SLC25A13基因所编码的citrin蛋白主要位于肝细胞、肾细胞及心肌细胞内,作为线粒体内膜上的天冬氨酸/谷氨酸载体,主要功能是将天冬氨酸由线粒体转运到细胞质内,同时将细胞质内的谷氨酸和质子转运到线粒体内,协同瓜氨酸参与尿素合成。因此,当citrin蛋白缺乏时,细胞质内天冬氨酸减少、瓜氨酸蓄积,尿素生成受阻,还原性辅酶I(NADH)与氧化型辅酶I(NAD+)比例升高,从而导致脂肪合成增加、糖酵解及糖异生减少等代谢紊乱。
瓜氨酸血症分为瓜氨酸血症Ⅰ型(citrullinemia,Cit-Ⅰ) 和瓜氨酸血症Ⅱ型,均属于常染色体隐性遗传的尿素循环障碍性疾病。瓜氨酸血症Ⅰ型是由于精氨酸琥珀酸合成酶(argininosuccinate synthetase,ASS) 基因突变所致,ASS1 基因(定位在 9p34.11) 突变使酶的功能缺陷,导致氨在体内蓄积,出现高氨血症,瓜氨酸及其他尿素循环的副产物在血液、尿液及脑脊液中蓄积,引起一系列的毒性损害,造成惊厥甚至昏迷等一系列临床表征,严重时导致脑水肿危及生命。瓜氨酸血症Ⅱ型是由于编码希特林蛋白(Citrin) 的 SLC25A13 基因(定位在 7q21.3)突变所致,引起尿素循环及 NADH 的转运障碍和相关代谢紊乱。成人发病患者称为成人发作的瓜氨酸血症Ⅱ型(adult onset type Ⅱcitrullinemia,CTLN2),1 岁内发病则称为 citrin 缺陷导致的新生儿肝内胆汁淤积症(neonatal intrahepatic cholestasis caused by citrin deficiency, NICCD),二者为 Citrin 缺乏症不同年龄的两种不同表型。
成人II型瓜氨酸血症(adult-onset type II citrullinemia,CTLN2)临床异质性大,新生儿至成人均可发病。新生儿期发病的患者表现为新生儿肝内胆汁淤积症,大多数患儿在生后第1年经对症治疗后可治愈,仅少数患儿进展为CTLN2。该病患者常于11-79岁发病,主要表现为发作性精神行为异常、抽搐、意识障碍,病情进展迅速者可致脑水肿和死亡。肝脏B型超声或活体组织检查可提示非酒精性脂肪肝(NAFLD)或肝硬化,严重者可发展至肝细胞癌。因此,早期诊断及治疗尤为重要,可明显改善患者的预后。
病例1
29岁的男子曾因新生儿黄疸住院,但原因不明,病情自行解决。他继续有不明原因的肝脏问题,直到他上初中,并接受熊去氧胆酸治疗。2016年因不明原因的肝损伤被转诊到另一家医院做彻底检查。各种病毒性肝炎测试和自身抗体测试均为阴性,并进行了肝活检,但病因仍不清楚。患者接受熊去氧胆酸和甘草甜素治疗。2020年3月进行的Gd-EOB-DTPA增强磁共振成像(EOB-MRI)显示S6和S8中的肝肿大和低血管结节,因此患者于2020年6月被转诊至我院进行详细检查。肝活检和核磁共振成像显示没有肝脂肪变性的证据。他的身高是168厘米,体重是58公斤;他神志清醒,眼结膜无黄化,胸部或腹部无异常发现。四肢没有明显的运动障碍。他的职业是建筑。他没有饮酒史。他母亲怀孕或分娩时没有并发症;他出生时的体重是2670克,身高是50厘米。如上所述,他曾因新生儿黄疸住院。此外,他在14岁时接受了阑尾切除术,没有围手术期问题。他没有急性胰腺炎病史。关于他的家族史,他与妻子和女儿住在一起。他的四个兄弟姐妹都没有经历过新生儿黄疸或随后的肝病。他的父母或其他亲属没有报告过肝病,他的父母没有血缘关系。
血液检查显示,他的肝胆酶、肾功能和电解质在标准范围内,但血氨升高。EOB-MRI显示肝脏增大,并且在S6显示尺寸为8 mm的低血管结节,在S8显示尺寸为5 mm的低血管结节,并且没有早期对比效应(图1)。腹部超声显示肝脏边缘变钝,内部回声粗糙,但没有显示肝脏脂肪变性的证据,如明亮的肝脏。纤维扫描值为8.3 kPa,CAP值为141.0 dB/m。b超和使用sonazoid的超声造影增强成像未显示病变。因此,几乎没有发现提示我们积极怀疑恶性肿瘤,我们将其诊断为再生或发育异常结节。因此,我们每六个月对患者进行一次EOB核磁共振随访。
经过介绍,他的详细生活史和病史显示,他从小就避免吃碳水化合物,如米饭和乌冬面,因为他吃这些东西时会呕吐,而且他有特殊的饮食史,喜欢吃蛋白质,如油炸食品、鱼和奶酪。他也不喝果汁,更喜欢喝牛奶,也不经常喝酒。他从童年开始就打棒球和踢足球,没有低血糖症状。25岁时,他因盗窃入狱。他的智力水平从童年起就被认为在正常范围内,并且在他犯罪时没有表现出任何神经精神症状。因此,他的监禁与神经精神症状无关。在监禁期间,他反复表现出异常行为,如夜间游荡、意识障碍、定向障碍和大小便失禁。当时医生对他进行了检查,但病因一直没有确定。
出狱后,他去看了精神病医生,医生诊断他患有惊恐障碍,并暂时用中草药治疗。然而,他随后根据自己的判断停止服用草药。出狱后,他的精神症状自然消失了。根据上述情况,怀疑CTLN2,血浆氨基酸分析显示瓜氨酸水平高达71.5 nmol/mL(表2)。为了确认诊断,在向患者及其家人充分解释了意义和细节后,进行了基因检测,发现导致瓜氨酸缺乏的基因SLC25A13以复合杂合的方式发生了病理突变(外显子9,c.852 _ 855del内含子11,c.1180+ 1G>A),这导致了CTLN2的明确诊断。父母没有CTLN2病史,也没有近亲结婚。他的四个兄弟姐妹也没有任何与CTLN2有关的病史;因此,没有进行基因检测。
病例2
患者男性,23岁,在读大学生,主因“反复精神行为异常1年余”于2015年12月9日入我院。患者于2014年7月开始无明显诱因反复出现精神行为异常,表现为躁动不安、胡言乱语、脱衣服等异常行为,发作前常有失眠、双手不自主震颤等症状,发病1个月余查血生化提示:ALT 112U/L(正常值1-40U/L),AST 89U/L(正常值1-37U/L),总胆红素39.2μmol/L(正常值2.0-20.0μmol/L),直接胆红素17.00μmol/L(正常值1.71-7.00μmol/L),血清铜7.5μmol/L(正常值11.0-24.0μmol/L),脑脊液常规、脑脊液生化、乙型肝炎两对半(乙型肝炎表面抗原、乙型肝炎表面抗体、乙型肝炎e抗原、乙型肝炎e抗体、乙型肝炎核心抗体)、头颅CT平扫、甲状腺功能等检查未见异常。
2014年9月查血生化提示:ALT 148U/L,AST 120U/L,血清铜蓝蛋白114mg/L(正常值150-300mg/L),血清铜9.0μmol/L(正常值11.0-24.0μmol/L)。头颅MRI提示双侧侧脑室前后角旁异常信号灶,考虑白质脱髓鞘改变。肝脏B型超声提示肝实质回声增高,上腹部CT提示脂肪肝,门静脉、胆囊、脾脏及肾脏等检查均未见异常。24h动态脑电图提示偶见大慢波呈节律性或长时程发放,外院诊断为“肝豆状核变性”,予以青霉胺125mg口服,每日3次驱铜治疗9个月后,上述症状仍反复发作。驱铜治疗期间多次复查血生化,ALT45-71U/L,AST40-90U/L,并先后4次复查血清铜蓝蛋白,分别为221、170、208、175mg/L。
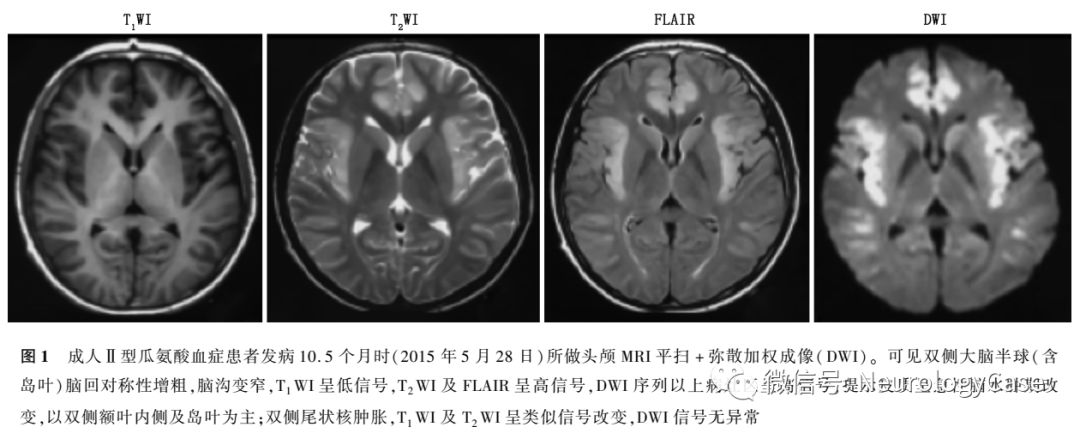
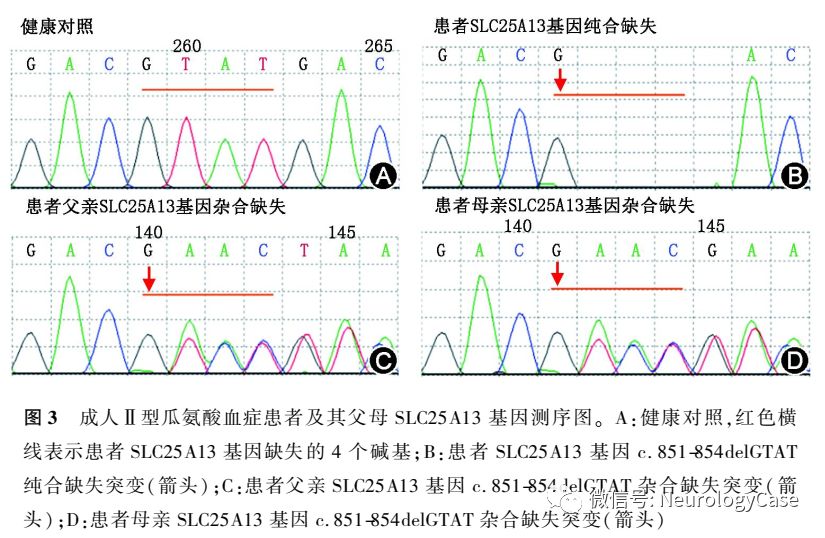
2015年5月10日患者再次无明显诱因出现发作性精神行为异常,外院仍考虑为“肝豆状核变性”,予以驱铜、护肝、营养神经等治疗7d后好转并出院。出院4d后再次出现精神行为异常,伴高热、视力模糊、意识障碍,体检呈深昏迷状态,查血氨363.4μmol/L(正常值9.0-33.0μmol/L),头颅MRI提示双侧大脑半球皮质呈急性脑水肿期改变,以双侧额叶内侧及岛叶为主;双侧尾状核异常信号改变(图1)。我院会诊后怀疑“瓜氨酸血症”,查血瓜氨酸568.2μmol/L(正常值5.5-45.0μmol/L),并采集患者外周血进行ATP7B及SLC25A13基因测序,结果提示SLC25A13c.851-854delGTATp.(Met285fs)纯合致病突变,诊断为“成人II型瓜氨酸血症”,予以降血氨(具体不详)治疗2周后,上述症状缓解,查血氨55.9μmol/L,遂办理出院。出院后患者自行停药,改服中药治疗(具体不详)。偶有胡言乱语等症状发作,次日可自行恢复正常,未予特殊处理,未定期监测血氨。
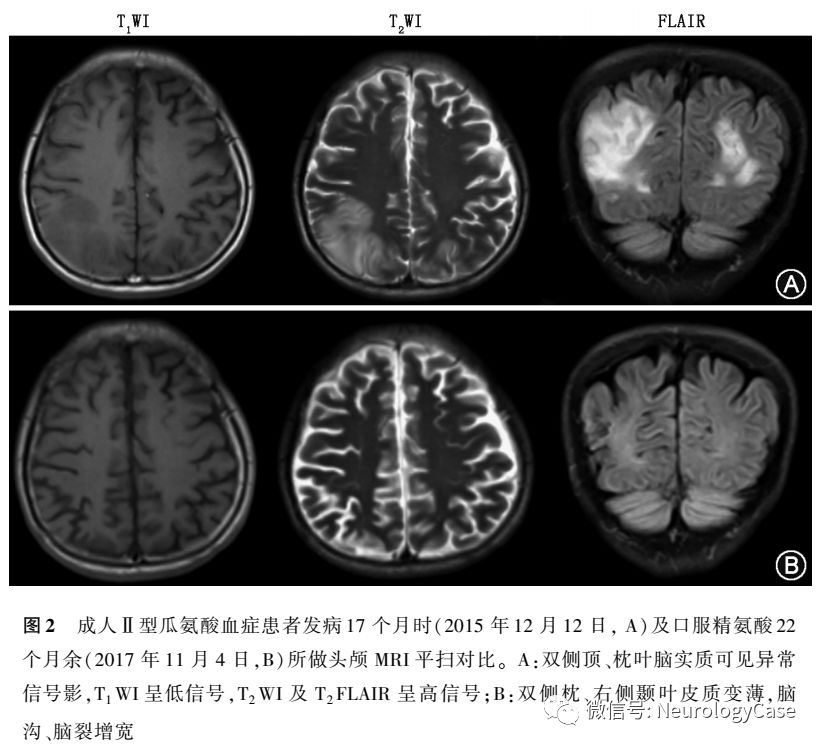
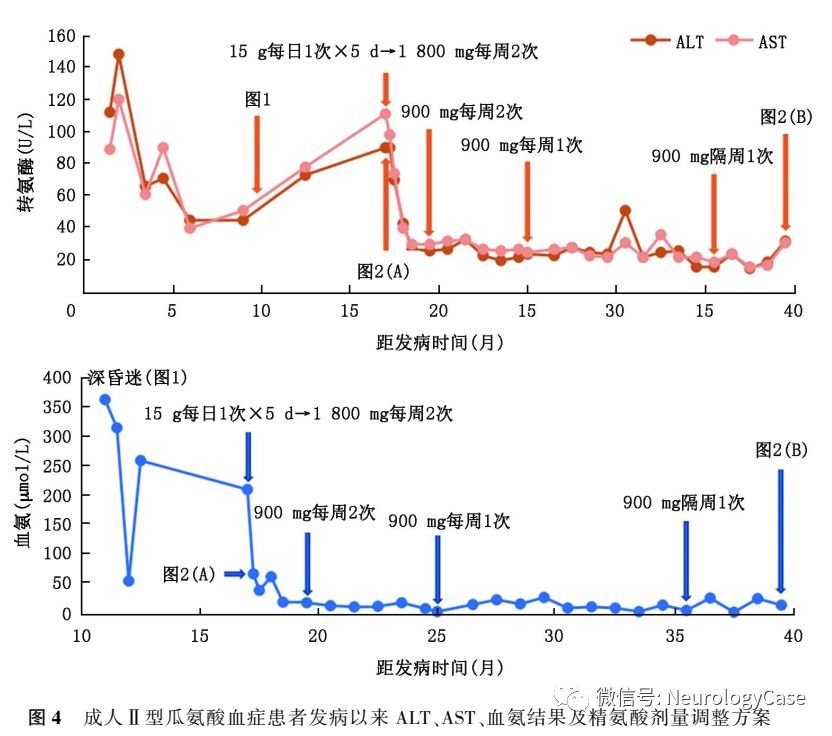
2015年12月患者为进一步治疗至我院就诊,自诉近1年来体重减轻约3kg。既往有转氨酶升高病史10余年,未予重视。平素挑食,喜食肉类、豆类、坚果类食物,形体消瘦。父母非近亲结婚,但来自邻近的乡镇。神经系统检查:远、近记忆力及计算力明显减退;双侧关节运动觉、图形觉减退,左下肢踝背屈肌力Ⅳ+级,双上肢Rossolimo征(+),四肢腱反射亢进,双侧踝阵挛阳性。MMSE评分为16分,查血生化提示ALT 90U/L,AST 111U/L,甘油三酯3.89mmol/L(正常值0.33-1.70mmol/L),血氨211μmol/L,血清铜蓝蛋白14.0mg/dl,考虑为“成人II型瓜氨酸血症”,予以精氨酸15g+0.9%氯化钠溶液500ml每日1次静脉滴注降血氨治疗,并嘱患者高蛋白高脂肪低碳水化合物饮食。入院3d后复查ALT 76U/L,AST 76U/L,血氨54μmol/L,头颅MRI提示双侧额、枕叶脑实质及双侧侧脑室旁脑白质异常信号,T1WI呈低信号,T2WI及T2 FLAIR呈高信号(图2)。入院5d后再次复查ALT 90U/L,AST 98U/L,血氨68μmol/L,患者父母基因检测结果回报均检测到携带SLC25A13c.851-854delGTATp.(Met285fs)突变(图3)。患者及其家属决定暂不行肝移植手术,入院5d后出院,出院后继续予以精氨酸降血氨治疗,并嘱患者高蛋白高脂肪低碳水化合物饮食。
随访:患者出院后坚持口服精氨酸1800mg,每周2次降血氨治疗,每月复查转氨酶及血氨,发病16.5个月后这2个指标均下降至正常范围内,并维持至今(图4)。发病20个月余复查血生化提示ALT 26U/L,AST 30U/L,血氨19μmol/L,遂遵医嘱调整精氨酸剂量至900mg,每周2次。发病25个月复查血生化提示ALT 24U/L,AST 25U/L,血氨4μmol/L。再次遵医嘱调整精氨酸剂量至900mg,每周1次。发病35.5个月复查血生化提示ALT16U/L,AST19U/L,血氨6μmol/L,遂第3次遵医嘱调整精氨酸剂量至900mg,2周1次至今。发病38个月患者返院复诊,未诉特殊不适,体检可见意识清楚,高级神经活动正常,双侧脑神经、四肢肌力、肌张力、感觉系统、共济运动等检查未见异常,双侧病理征阴性。MMSE评分为30分,复查血生化提示ALT32U/L,AST31U/L,血氨15μmol/L,血瓜氨酸155.63μmol/L,肝脏B型超声提示正常,头颅MRI提示双侧枕、右侧颞叶软化灶及皮质较前变薄(图2)。
延伸阅读
瓜氨酸血症Ⅱ型是由于编码希特林蛋白(Citrin) 的 SLC25A13 基因(定位在 7q21.3)突变导致的常染色体隐性遗传病。希特林蛋白的功能主要是作为线粒体内膜上天冬氨酸 / 谷氨酸的载体,参与一系列的生物反应过程,将线粒体内天冬氨酸与胞质中谷氨酸、质子交换,向胞质提供天冬氨酸参与尿素、蛋白质和核苷酸合成。同时,作为苹果酸 / 天冬氨酸穿梭的一员,将胞质中还原型烟酰胺腺嘌呤二核苷酸(NADH) 运至线粒体,向线粒体内提供充足的 NADH,同时维持线粒体与胞质之间 NADH 比例的平衡。SLC25A13 基因突变可影响希特林蛋白的活性,由线粒体转运到胞质的天冬氨酸减少,导致胞质内天冬氨酸缺乏,尿素循环受阻、瓜氨酸蓄积;同时导致胞质内 NADH/NAD+ 升高,从而引发各种代谢紊乱如抑制糖酵解、糖异生,干扰蛋白质及核酸合成,同时抑制脂肪酸氧化、促进脂肪合成。
瓜氨酸血症Ⅱ型在亚洲、北美和欧洲均有报道,而患者主要分布在东亚地区。在日本人群中,SLC25A13 基因纯合子突变频率约 1/19 000,与 NICCD 发病率一致,CTLN2 的发病率为 1/230 000~1/100 000。有报道估计在我国南方 SLC25A13 基因纯合子突变频率约1/9 200,而在我国北方约 1/3 500 000,南方比北方的发病率高。
对于转氨酶及血氨升高的青年非妊娠期患者,需排除感染性、中毒性、药物性、酒精性、肿瘤性等病因,以及上消化道出血、感染、高蛋白饮食、肾功能不全等常见诱因。在排除上述病因及诱因的前提下,结合该患者发作性精神行为异常、转氨酶及血氨升高、喜食富含蛋白或脂肪的食物等临床特征,可高度怀疑CTLN2。高达89%的CTLN2患者伴有NAFLD,其中涉及细胞质内脂肪酸的过度生产、摄入以及氧化、分泌、转运抑制。若患者同时伴有胰腺炎、体重指数小于20kg/m2、血清胰蛋白酶抑制物大于29ng/ml等临床特征,则进一步支持CTLN2的诊断。
NICCD 患儿于出生后至数月内发病。发病年龄多在 2 月龄内,很少晚于 5 月龄。最常表现为出生后黄疸持续不退伴或不伴大便颜色浅淡,肝脏轻度肿大或无明显肿大。NICCD 还有其他多种非特异临床表现,包括喂养困难、生长发育迟缓、腹泻、低血糖、凝血时间延长等。大多数 NICCD 患者上述症状及体征在 6 个月至 1 岁左右可自然缓解或经过饮食结构调整和药物治疗缓解,其中少部分的 NICCD 患者在青春期以后发展为 CTLN2。
CTLN2 大部分是在成人后发病。通常在饮酒、摄入甜食、服用某些药物或感染后突然出现高氨血症所致的谵妄,意识混乱等精神异常表现,严重者易与肝性脑昏迷混淆。严重的 CTLN2 病情发展急剧,得不到正确治疗可危及生命。患者多为消瘦体形,大部分患者有明显的饮食偏好,嗜食豆类、高蛋白、高脂食物,而厌食高糖类。
目前国内外尚无公认的 NICCD 临床、生化诊断标准。1 岁以内起病,临床以黄疸为主要表现,血生化提示血清总胆红素和直接胆红素升高,血瓜氨酸等多氨基酸升高的患者应考虑 NICCD 的可能,结合氨基酸升高的患者应考虑 NICCD 的可能,结合 SLC25A13 基因突变分析被认为是确诊NICCD 的可靠手段。患者的两个 SLC25A13 等位基因均有致病性突变即可确诊本病。
目前尚无有关 CTLN2 的诊疗指南。CTLN2 的诊断可参考以下几点:①喜食富含豆类、高蛋白质和高脂的食物,厌食富含糖类的食物;②一般营养状况差,BMI 明显偏低;③临床表现为发作性神经精神系统症状;④实验室检查显示高瓜氨酸血症和高氨血症;⑤肝活组织检查常表现为脂肪肝;⑥基因检测为 SLC25A13 位点突变。
对于 NICCD 患儿,除了与婴儿肝炎综合征中感染相关的疾病及肝外胆道梗阻(如先天性胆道闭锁、胆总管囊肿) 相鉴别外,还需要与其他引起肝内胆汁淤积的疾病加以鉴别;对于 CTLN2,需与各种原因肝硬化所导致的肝性脑病、瓜氨酸血症Ⅰ型等疾病相鉴别。
治疗方法及疗效
NICCD 的治疗 主要包括饮食调整和对症治疗。NICCD 患儿需改用无乳糖配方奶和(或) 强化中链三酰甘油(MCT) 的治疗奶粉。同时补充脂溶性维生素(包括维生素 A、D、E、K)。熊去氧胆酸可用于利胆。大部分患者预后良好,症状可在 1 岁内缓解,个别患者预后不良。
CTLN2 的治疗包括:
(1) 饮食治疗:适当降低糖类在总热量的占比、适当提高蛋白和脂类的占比;如血氨明显升高者,则需要控制蛋白质的摄入量。
(2) 补充丙酮酸:丙酮酸钠可通过乳酸脱氢酶反应将细胞质内的 NADH 氧化为NAD+,为三羧酸循环的底物提供能量,同时改善 Citrin 缺乏所导致的氧化应激。有个案报道显示口服丙酮酸钠(4.5~8g/d) 可减少 CTLN2 患者高氨血症发作。
(3) 脑病发作时治疗:由 CTLN2 的代谢特点可知,饮酒、含糖食物及某些药物可诱发或加重 CTLN2 患者的临床症状。常规治疗肝性脑病的方法,如高糖类、低蛋白饮食以及输注富含高糖的溶液,均不能用于 CTLN2 患者的治疗。精氨酸能改善高氨血症和瓜氨酸血症,可能对治疗 CTLN2 有效。
(4) 肝脏移植:肝移植是目前公认对本病最有效的治疗方法。可以预防高氨血症导致的相关脑病出现,纠正代谢紊乱,改善嗜好高蛋白的饮食习惯。
目前公认肝移植仍是CTLN2最有效的治疗手段,可缓解肝细胞内尿素循环异常所致的代谢紊乱,从而防止高氨血症、改善高蛋白高脂肪低碳水化合物的饮食偏好。日本超过40例接受肝移植的CTLN2患者存活率达到95.6%。由于肝源极度匮乏,自1996年Yazaki成功进行第1例CTLN2患者的活体亲属部分肝移植术(living related partial liver transplantation,LRLT),大部分学者推荐使用SLC25A13基因突变携带者的肝脏作为供体进行LRLT,例如CTLN2患者的父母或直系兄弟姐妹等。然而,肝移植高昂的手术费用仍使众多患者选择放弃该治疗方法。
CTLN2是一种以高氨血症和血清瓜氨酸水平升高为特征的代谢性疾病,主要表现为消瘦和反复发作的意识障碍,并表现出对富含蛋白质和脂质的食物的饮食偏好。多由Citrin缺乏引起。Citrin是一种天冬氨酸-谷氨酸盐载体,与肝脏能量供应密切相关。Citrin缺乏症是由染色体7q21.3上的SLC25A13基因突变引起的,该突变可导致新生儿肝内胆汁淤积症(NICCD)或CTLN2。肝移植被认为是治疗CTLN2最有效的方法。然而,患者不良的术前营养状况显著增加了移植后并发症的发生率、发病率和死亡率。
自2003年起,药物治疗CTLN2的成功率大大提升,其中疗效最好的是精氨酸和丙酮酸钠。丙酮酸钠被发现可降低肝细胞质内NADH/NAD+比例,促进尿素生成。在Citrin基因敲除小鼠或Citrin/线粒体三磷酸甘油脱氢酶基因双敲除小鼠中,乳酸/丙酮酸比例升高导致细胞质内NADH/NAD+比例随之升高,外源性丙酮酸盐能纠正乳酸/丙酮酸比例,从而诱导细胞质内NADH再氧化。采用“CTLN2”在万方数据库、CNKI数据库及PubMed数据库中检索,1996年至今国内外共有26篇文献报道利用LRLT、精氨酸或丙酮酸钠治疗CTLN2,随访时间从2周到6年不等,其中18例接受LRLT及3例接受精氨酸+丙酮酸钠治疗的患者存活率达到100%。6例接受精氨酸治疗的CTLN2患者中有1例死亡,其余均存活(表1)。该死亡病例除精氨酸治疗外,同时予以支链氨基酸(branched chain amino acids,BCAA)、甘油、乳果糖、卡那霉素及低蛋白饮食治疗。然而,静脉输注BCAA、甘油及低蛋白饮食治疗被报道可加重脑水肿程度:BCAA可能干扰尿素循环;甘油会增加细胞质内NADH/NAD+比例;为保证足够的能量摄入,低蛋白饮食必然会导致高糖饮食,加之稀释BCAA及甘油所需的葡萄糖注射液,机体内会快速产生大量NADH,这些都可能是该病例的死亡原因。
对于反复发作性精神行为异常、转氨酶及血氨升高的患者,需考虑CTLN2。当患者有转氨酶反复升高、NAFLD及体重指数<20kg/m2而无精神症状时,排除感染性、药物性及中毒性等病因后,也需高度警惕CTLN2。LRLT、精氨酸或精氨酸+丙酮酸钠是目前针对CTLN2最有效的治疗方法,经济条件允许的情况下,早期进行肝移植可明显改善高氨血症及瓜氨酸血症,且不需进行饮食限制。早期、足量、连续服用精氨酸亦可有效改善预后,与丙酮酸钠联用可加强降血氨疗效,治疗期间需严格遵循高蛋白高脂肪低碳水化合物的饮食要求,并定期监测血气分析及电解质,防止高氯性酸中毒及高钾血症。最后,临床医生应加强对CTLN2患者及其家属的宣传教育及遗传咨询工作,早期诊断及正确治疗能明显改善该病的预后,提高患者的生活及工作质量。
参考文献
Okano Y, Okamoto M, Yazaki M, Inui A, Ohura T, Murayama K, Watanabe Y, Tokuhara D, Takeshima Y. Analysis of daily energy, protein, fat, and carbohydrate intake in citrin-deficient patients: Towards prevention of adult-onset type II citrullinemia. Mol Genet Metab. 2021 May;133(1):63-70.
尤桦菁,吴超,李洵桦.成人Ⅱ型瓜氨酸血症一例临床特征、治疗及随访分析[J].中华神经科杂志,2018,51(07):533-539.
Suzuki T, Matsuura K, Imura N, Kawamura H, Kuno K, Fujiwara K, Nojiri S, Ito S, Togawa T, Kataoka H. Adult-onset Type II Citrullinemia Developed under Dietary Restrictions during Imprisonment. Intern Med. 2024 Mar 15;63(6):833-837.
Du Y, Fu YY, Yue Y, Han B, Zhang WJ, Yu DC, Bian XJ. Nutritional support therapy for liver transplantation in an adult-onset type II citrullinemia patient: a case report. Front Nutr. 2024 Mar 22;11:1364866.

