特别关注|胰源性糖尿病的发病机制探析
时间:2024-09-22 06:00:40 热度:37.1℃ 作者:网络
2014年美国糖尿病学会将由胰腺外分泌疾病导致的糖尿病命名为胰源性糖尿病或3c型糖尿病(T3cDM)。国外有研究显示胰源性糖尿病的患病率占所有糖尿病患者的5%~10%,但由于缺乏系统的流行病学调查,故真正的患病率目前相对未知。有调查表明胰源性糖尿病的患病率显著高于1型糖尿病,但仍低于2型糖尿病。慢性胰腺炎是胰源性糖尿病最常见的病因,约占80%,其次是胰腺癌,约占8%。因各种原因导致的胰腺β细胞或α细胞缺乏使得胰源性糖尿病患者的血糖尤难控制,因此胰源性糖尿病又被称为“脆性糖尿病”,更容易发生与低血糖相关的并发症,甚至死亡。国外研究表明,胰源性糖尿病患者比2型糖尿病患者有更高的死亡和再入院风险,并且有文献表明,这种由胰腺外分泌疾病引起的糖尿病误诊率极高,45%~90%的胰源性糖尿病病例被错误地归类为2型糖尿病。目前国内对胰源性糖尿病的研究及报道较少,具体发病率和发病机制尚不完全明确,因此充分了解胰源性糖尿病的发病机制,对临床研究该疾病以提高其诊断及治疗水准,改善患者的生存及生活质量具有重要意义。
2021年美国糖尿病协会制定的胰源性糖尿病诊断标准为:(1)符合糖尿病诊断标准,(2)存在胰腺分泌功能不全,(3)影像学表明胰腺病理性改变,(4)不具有1型糖尿病相关的自身免疫标志物。胰源性糖尿病的发生可能与以下机制相关。
1免疫介导下的胰岛功能障碍
免疫介导下的胰腺纤维化和胰岛功能障碍是胰源性糖尿病发生的关键机制。在慢性胰腺炎和胰腺癌环境中,高水平的细胞因子和应激性炎症环境引起胰岛β细胞功能障碍,之后更多的免疫细胞被募集到胰腺组织中损伤胰腺腺泡细胞,随后胰腺星状细胞被激活,并与各种免疫细胞相互作用,进一步加重胰腺组织的纤维化。由于胰腺实质的严重纤维化,胰腺腺泡、导管和神经束等有功能的胰腺组织最终被结缔组织取代,从而失去血管及分泌功能,进而促进了糖尿病的发生(图1)。
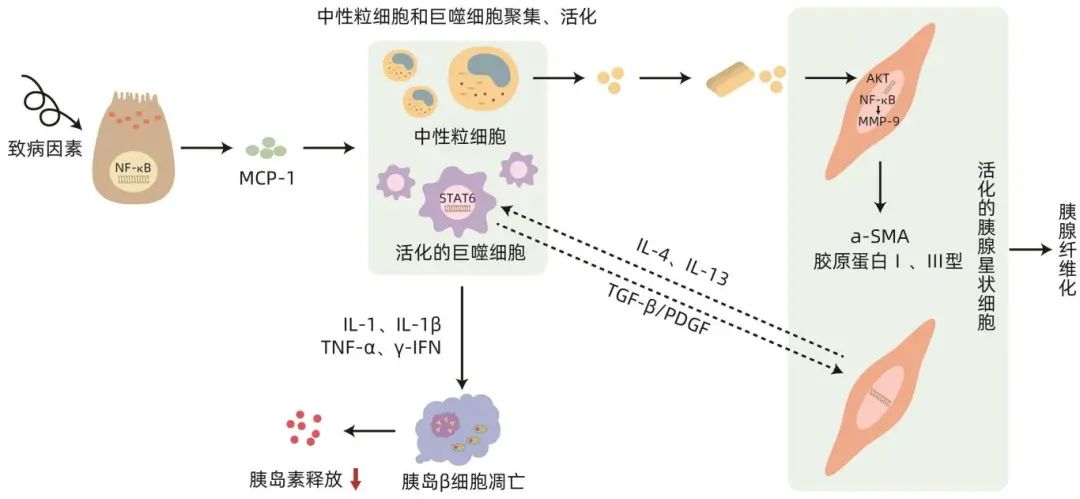
图1 免疫机制引发胰腺纤维化示意图
1.1 巨噬细胞
胰源性糖尿病的发生发展与巨噬细胞的募集和活化引发炎症细胞大量释放进而导致胰岛功能改变相关。Li等研究表明,巨噬细胞的募集和活化是加重胰腺细胞损坏的重要环节之一。巨噬细胞是胰腺炎发展过程中浸润胰腺的主要炎症细胞,在胰腺纤维化进展中起重要作用。胰腺中的巨噬细胞部分来源于胚胎中的卵黄囊祖细胞,可通过骨髓造血干细胞来源的单核细胞的分化来补充。巨噬细胞可分为M1型和M2型。M1型巨噬细胞可能通过产生促炎细胞因子和特异性趋化因子来诱导Th1型细胞免疫反应,这些趋化因子介导宿主对多种细菌、寄生虫和病毒的防御;M2型巨噬细胞参与抗寄生虫功能、组织修复和重建、肿瘤血管生成和免疫调节。胰腺炎发生机制复杂,胰腺中巨噬细胞的募集和活化及其与胰腺腺泡细胞的相互作用,促进了该病的发生和发展。多种病理刺激导致胰腺腺泡细胞的损伤,这些受损的胰腺腺泡细胞将胰蛋白酶、细胞因子、炎症介质和其他细胞内容物释放到血液中,又进一步引发胰蛋白酶原释放和NF-κB炎症信号通路激活。NF-κB信号通路激活后,腺泡细胞会分泌单核细胞趋化蛋白-1(MCP-1)来募集CCR2受体阳性的炎性单核细胞或巨噬细胞。然后,这些募集的单核细胞或巨噬细胞联合组织中的巨噬细胞群将更多的炎症介质、细胞因子和生长因子分泌到局部的炎症环境中,这反过来又会吸引更多的免疫细胞群进入胰腺,从而进行表型和功能的变化,放大局部炎症并导致炎症级联反应。肿瘤相关巨噬细胞(TAM)是浸润肿瘤组织或聚集在实体肿瘤微环境中的巨噬细胞。在胰腺癌早期,TAM会表现出更多的促炎M1表型,促进抗肿瘤活性,而随着疾病的进展,则会表现出更多的M2表型。M1促炎谱表达的最重要的细胞因子之一是TNF-α,而抗炎表型M2产生免疫抑制细胞因子IL-10。这些促炎细胞因子如IL-1β、TNF-α、IFN-γ可抑制受葡萄糖刺激的胰岛素释放,高浓度IL-1受体(IL-1R)和IL-1β可诱导β细胞凋亡,而M2巨噬细胞可与胰腺星状细胞相互作用,加重胰腺组织的纤维化,进而增加继发糖尿病的风险。
1.2 中性粒细胞
中性粒细胞通常是募集到炎症部位的第一个效应细胞,在先天免疫反应中起着重要作用。中性粒细胞不仅可以产生大量的活性氧和活性氮,导致炎症恶化,还可以激活各种免疫细胞和基质细胞分泌炎性细胞因子,导致炎症加重。在充分刺激下,中性粒细胞可释放过量的活性氧,进而诱发细胞和组织损伤。在胰腺炎中,活性氧可以激活胰腺星状细胞中下游的氧化还原敏感信号转导通路AKT和NF-ĸB,上调MMP-9和Twist的表达,产生α-SMA及胶原Ⅰ和Ⅲ,促进胰腺细胞的纤维化,加速胰腺炎后糖尿病的发生。
1.3 T淋巴细胞
最近的研究表明,慢性胰腺炎与疾病特异性调节性T淋巴细胞反应有关,这些调节性T淋巴细胞在血液、骨髓和胰腺炎病灶中扩增,具有以抗原特异性方式抑制自体常规T淋巴细胞增殖的潜力。与胰腺癌相比,胰腺炎中的IL-10升高,IFN-γ水平降低,表明原位调节性T淋巴细胞具有胰腺炎特异性活性。有研究还发现急性胰腺炎期间外周血中CD4+T淋巴细胞会迁移到炎症部位,导致胰腺中CD4+T淋巴细胞计数显著增加,慢性胰腺炎外周血中CD3+T淋巴细胞群和循环CD4+T淋巴细胞群显著增加。这些发现表明,T淋巴细胞也可能在胰腺炎的进展中发挥重要作用。另有研究表明Treg细胞能够释放IL-4/IL-13抑制Th2细胞分化来控制胰腺巨噬细胞,通过抑制巨噬细胞反应和生长因子释放,防止慢性胰腺炎过程中胰腺组织过度纤维化。
1.4 胰腺星状细胞
胰腺星状细胞的激活程度决定了胰腺纤维化的严重程度。在功能上,胰腺星状细胞是位于腺泡细胞、小血管和胰管周围的多能效应细胞。在健康的胰腺组织中胰腺星状细胞处于静止状态,数量很少,仅占胰腺细胞总数的4%~7%。纤维化是胰腺癌和慢性胰腺炎的共同特征,胰腺星状细胞负责促进和维持纤维化。在胰腺炎中,胰腺星状细胞被浸润在胰腺周围的免疫细胞释放的促炎物质激活,引发肌成纤维细胞样表型。特别是活化的胰腺星状细胞高度表达a-SMA、ECM、Ⅰ型和Ⅲ型胶原以及纤连蛋白,逐渐取代健康的胰腺组织,最终导致胰腺纤维化。巨噬细胞还与胰腺星状细胞相互作用,促进慢性胰腺炎的进展。胰腺炎的致病因素会对腺泡细胞造成损害,腺泡细胞通过募集和激活炎性单核细胞与巨噬细胞,释放IL-1、IL-6、TNF-α、血小板源性生长因子(PDGF)、TGF-β1、激活素A、乙醇及其代谢物等,这些细胞因子可作为胰腺星状细胞激活的调节剂。然后,活化的星状细胞释放IL-4/IL-13,诱导M2巨噬细胞的分化。相应地,M2巨噬细胞也可以释放TGF-β/PDGF来激活星状细胞,进一步分泌a-SMA/ECM引起胰腺纤维化。胰腺癌的基质主要由胰腺星状细胞铺设的胶原纤维(占其体积50%~80%),以及其他细胞和非细胞成分组成,这些基质成分可以介导胰腺星状细胞与癌细胞之间的相互作用。胰腺星状细胞可促进癌细胞增殖、迁移、侵袭、EMT和转移,还可以通过减少癌细胞凋亡来促进癌细胞存活。癌细胞反过来产生PDGF、三叶因子1(TFF1)和COX-2等因子,来诱导胰腺星状细胞增殖、收缩、迁移和增加胶原蛋白合成,从而使胰腺组织失去原本的分泌功能。
2胰岛素抵抗
胰岛素抵抗是糖尿病的一个突出特征,同时也可能是胰腺炎后继发糖尿病的一个重要因素。研究发现胰源性糖尿病患者均出现了肝胰岛素抵抗。有研究表明,胰多肽应答不足在肝胰岛素抵抗中起着关键作用。现已证实胰多肽是一种调节肝脏胰岛素敏感性的血糖调节激素,它可以逆转胰腺炎中胰岛素受体的减少。有发现表示,胰多肽可增强胰源性糖尿病患者的胰岛素敏感性,并减少胰岛素的使用量。另有动物实验研究表明,静脉注射胰多肽可逆转慢性胰腺炎大鼠的肝胰岛素抵抗。慢性胰腺炎患者肝胰岛素功能的改变除了与胰岛素受体可用性改变相关以外,也与肝细胞IκBβ和NF-κB的炎症激活有关,抑制NF-κB的活化可改善肝胰岛素的敏感性。胰腺癌患者同样具有胰岛素抵抗,研究发现胰腺癌患者的胰岛淀粉样蛋白多肽(IAPP)水平高于其他癌症、糖尿病或健康对照患者,这种物质已被证实会导致骨骼肌中的胰岛素抵抗。在慢性胰腺炎和胰腺癌中巨噬细胞释放的炎性细胞因子如TNF-α、IFN-γ和IL-6也会引起或促进胰岛素抵抗的发生。TNF-α可以通过激活IκBβ或NF-κB通路增加胰岛素受体底物-1(IRS-1)的丝氨酸/苏氨酸磷酸化来引起胰岛素抵抗。IFN-γ通过抑制脂肪细胞中的胰岛素作用和脂联素的分泌来促进胰岛素抵抗的发生。IL-6通过激活Janus激酶信号转导和转录激活因子(JAK/STAT)信号通路及诱导细胞因子信号转导抑制因子3(SOCS3)的表达来降低GLUT4与IRS-1的表达,引起胰岛素抵抗。此外,IL-6还可以通过下调miR-200、miR-301a的表达来抑制糖原的合成,进一步诱导胰岛素抵抗的发生。
3促胰岛素效应减少
胰高血糖素样肽-1(GLP-1)和葡萄糖依赖性胰岛素生长多肽是肠道消化与吸收营养物质时释放的一种激素。GLP-1具有调节胰岛素分泌的功能,能刺激胰岛素的释放,抑制胰高血糖素的分泌,并增加胃排空的时间。此外,GLP-1还有促进胰腺β细胞生长、分化和保护β细胞的作用。胰岛素生长多肽可促进胰岛素释放而抑制α细胞分泌胰高血糖素。胰腺炎时一系列的炎症反应会破坏胰腺功能,导致营养物质吸收障碍,肠促胰岛素激素反应性的释放减少,进而引起餐后胰岛素分泌减少,血糖相应的升高。
4肠道菌群紊乱
现已发现肠道菌群在糖尿病的发生发展中具有独特的作用。肠道微生物群能够参与人体的代谢和炎症反应。研究发现,胰腺炎发生时的各种理化因素和致病因子引起的胰腺损伤和炎症反应会导致肠黏膜屏障完整性受损,从而促使肠道中的微生物群发生改变。肠道生态和细菌代谢的变化会反过来影响糖尿病与人体代谢。印度的一项研究招募了健康对照、1型糖尿病患者、2型糖尿病患者和继发于慢性胰腺炎的胰源性糖尿病患者,在4组中发现了某些细菌物种的丰度显著差异,包括瘤胃球菌属和粪杆菌门。另一项研究在未发生糖尿病的慢性胰腺炎患者中观察到粪杆菌丰度降低和血浆内毒素增加,这在继发了糖尿病的慢性胰腺炎患者中更加明显。该研究还发现空腹和餐后血糖与粪杆菌丰度呈显著负相关,而血浆胰岛素水平与粪杆菌丰度呈显著正相关,这提示肠道微生态与慢性胰腺炎代谢变化相关。综上,肠道微生物群可能在胰腺炎相关糖尿病的发生、发展中起到作用。
5遗传因素
研究已经确定了慢性胰腺炎的特异性基因,如PRSS1、SPINK1、CFTR、CTRC和CASR。CFTR主要在胰管内衬导管和中央腺泡细胞的顶端质膜上表达,并控制cAMP刺激的HCO3-分泌物进入导管腔,CFTR在胰腺中的主要功能是稀释和碱化富含蛋白质的腺泡分泌物,以防止蛋白栓的形成而损伤胰腺,故若CFTR发生突变便可能会促进胰腺纤维化的发生。在胰腺组织中PRSS1、SPINK1和CTRC仅在腺泡细胞中表达,这些基因若发生突变会导致腺泡内的胰蛋白酶原过早激活,从而引发胰腺损伤,加速胰腺组织的破坏和纤维化。研究表明PRSS1外显子区域的突变与慢性胰腺炎继发糖尿病有关。在慢性胰腺炎患者中,外显子异常有60%的概率发展为糖尿病。
6展望
随着胰腺炎和胰腺癌发病人群的不断增加,胰源性糖尿病的发病率也不断升高。但由于胰源性糖尿病概念提出的时间相对较短,导致现阶段国内普遍缺乏对该疾病系统的认识。故而该类型糖尿病常会被误诊为2型糖尿病。但与目前患病率较高的2型糖尿病相比,胰源性糖尿病患者的血糖控制更难,低血糖发作更频繁、胰岛素使用剂量更多、方案更复杂,发生癌症的风险更高,死亡年龄更早,死亡风险更大。所以及时正确地诊断胰源性糖尿病有重要意义,这能大大提高此类型糖尿病患者的生存和生活质量,并降低致残率和死亡率。遗憾的是,目前国内对该疾病的关注度尚低,也缺乏相关的流行病学数据,更未形成广泛认可的诊断、治疗和管理指南,这无疑给临床工作带来了巨大的挑战。期待未来有更多该方向的相关研究来解决目前临床存在的问题。
全文下载
https://www.lcgdbzz.org/cn/article/doi/10.12449/JCH240834


