eGastroenterology:PTPRD-STAT3轴:代谢性肝病治疗新靶点的突破性发现
时间:2025-02-08 12:08:53 热度:37.1℃ 作者:网络
导读
代谢相关脂肪性肝病(metabolic dysfunction-associated steatotic liver disease,MASLD)是指合并一种或多种心血管代谢危险因素且无过量酒精摄入的脂肪性肝病,是慢性肝病(chronic liver disease,CLD)的重要病因。代谢紊乱是其进展的核心驱动因素,但相关分子机制仍待阐明。近期,法国斯特拉斯堡大学研究团队联合法国里昂癌症研究中心、加拿大麦吉尔大学及美国德克萨斯大学西南医学中心的科研人员,在eGastroenterology期刊发表了题为“Protein tyrosine phosphatase delta is a STAT3-phosphatase and suppressor of metabolic liver disease”的原创性研究,首次揭示蛋白酪氨酸磷酸酶δ(protein tyrosine phosphatase delta,PTPRD)在肝脏糖脂代谢稳态中的关键调控作用。研究发现,PTPRD通过抑制信号转导及转录激活因子3(signal transducer and activator of transcription 3,STAT3)信号通路来维持肝脏代谢平衡,并证实其表达缺失与代谢性肝病的发生密切相关。这一突破性发现不仅加深了对代谢性肝病机制的理解,也为开发靶向PTPRD-STAT3轴的创新疗法提供了重要的理论依据。
PTPRD是受体型蛋白酪氨酸磷酸酶(RPTP)家族成员,其结构特征包含细胞外结构域、跨膜结构域及胞内催化结构域,通过催化底物蛋白的酪氨酸去磷酸化,调控细胞信号转导,在神经、肿瘤及代谢方面发挥调控作用。
STAT3是STAT蛋白家族成员,是一种兼具信号转导和转录调控双重功能的蛋白质,具有SH2结构域、DNA结合域和转录激活域,通过磷酸化激活及二聚化与核转位,响应细胞外信号,调控靶基因表达,参与细胞增殖、分化、凋亡及免疫调节等过程,在癌症、炎症与免疫及代谢方面扮演重要角色。
名 词 解 释
PTPRD驱动代谢紊乱的分子机制
研究团队通过分析GEO数据库发现,无CLD人群肝脏中PTPRD低表达可激活STAT3等炎症信号通路,抑制过氧化物酶体功能并扰乱糖脂代谢,这一现象与丙肝患者中PTPRD-STAT3轴的异常激活高度一致。通过进一步构建Ptprd缺陷小鼠模型发现,肝脏驻留细胞比例或免疫细胞浸润未被影响,但杂合小鼠(Ptprd+/-)有关糖尿病进展转录程序被活化,胰岛素信号通路被显著抑制,这证实PTPRD是肝脏代谢通路的调控因子。为探究PTPRD在病理条件下的作用,本研究还分析了不同病因CLD患者的肝脏转录组数据,发现代谢紊乱相关肝病(如MASLD、酒精性肝病、慢性丙型肝炎)PTPRD表达降低,丙肝显著,而乙肝未受影响,符合MASLD和丙肝患者较乙肝患者更易发生脂肪肝的临床特点。进一步单细胞测序分析发现,高脂高糖饮食小鼠及原发性硬化性胆管炎/原发性胆汁性胆管炎(PSC/PBC)患者的肝细胞中Ptprd表达显著降低,表明PTPRD与MASLD病理机制特异性相关。为明确其直接作用,研究者进一步利用胆碱缺乏高脂饮食(CDA-HFD)诱导小鼠MASLD模型,结果发现Ptprd杂合较野生型小鼠相比,虽然体重增长无差异,但出现空腹血糖升高、肝脏脂质沉积增加及脂肪酸谱异常(如棕榈酸积累、油酸和γ-亚麻酸水平减少),与2型糖尿病及MASH患者的代谢特征高度吻合。
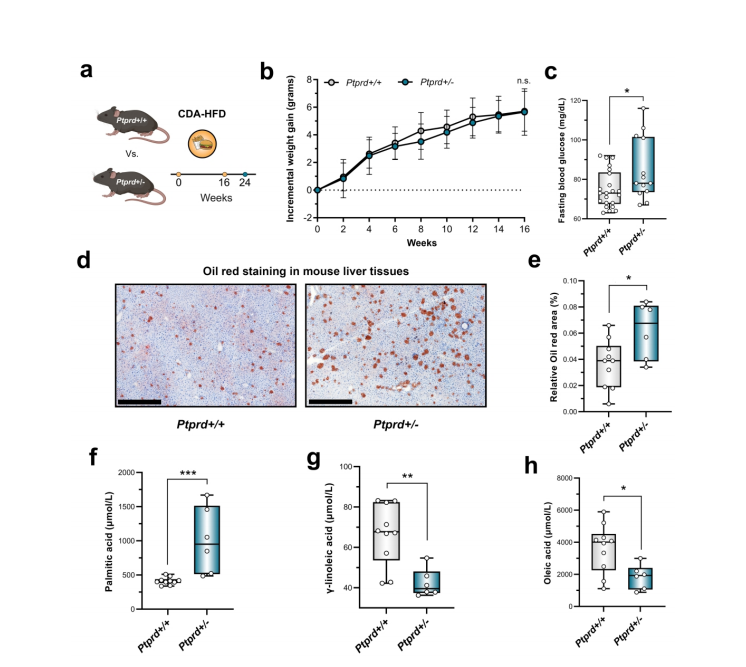
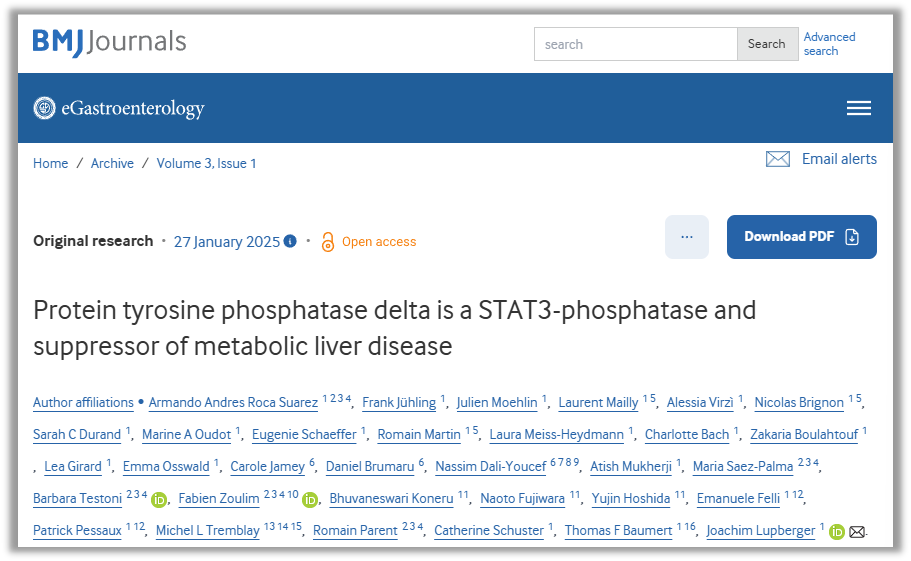
图1:Ptprd 缺陷促进代谢性疾病表型
a)Ptprd缺陷肝脏疾病小鼠模型。8周龄雄性Ptprd+/+(n=22)和Ptprd+/-(n=13)小鼠接受16周胆碱缺乏高脂饮食(CDA-HFD),随后转为正常饮食8周后处死。b)在16周的CDA-HFD干预期间,Ptprd+/-小鼠的体重增长趋势与Ptprd+/+小鼠相似。c)CDA-HFD干预8周后,Ptprd+/-小鼠的空腹血糖水平显著高于Ptprd+/+小鼠(T检验)。d-e)与Ptprd+/+小鼠(n=10)相比,Ptprd+/-小鼠(n=6)在CDA-HFD干预后肝脏脂质水平显著升高(U检验)。油红O染色显微镜分析结果(标尺500 µm)。f-h)Ptprd缺陷小鼠的肝脏脂质谱异常。CDA-HFD干预后,Ptprd+/-小鼠(n=6)的肝内棕榈酸、油酸和γ-亚麻酸水平与Ptprd+/+小鼠(n=10)相比发生显著改变(U检验)。数据以µmol/L表示(经总蛋白浓度标准化)。
注释:CDA-HFD,胆碱缺乏、L-氨基酸定义型高脂饮食;Ptprd,蛋白酪氨酸磷酸酶δ。*p<0.05,**p<0.01,***p<0.001,****p<0.0001。
来源:原文图3
为深入解析PTPRD与肝脏代谢的关联机制,本研究鉴定了人类肝脏微环境中表达PTPRD的细胞群体。结果发现在人类肝脏中,PTPRD主要富集于肝上皮细胞(肝细胞、胆管细胞及肝祖细胞(liver progenitor cells,LPCs)),其中双能LPCs表达水平最高。另外,单细胞分析显示,PTPRD水平与STAT3靶基因表达呈显著负相关(r=-0.46),且Ptprd缺陷小鼠肝脏中STAT3磷酸化水平呈等位依赖性升高,这表明PTPRD直接抑制STAT3活性。底物捕获实验进一步证实,重组PTPRD-GST融合蛋白可特异性结合磷酸化STAT3,而基因敲低PTPRD催化域则可增强IL-6诱导的STAT3活化,证实STAT3是PTPRD底物。在进一步过氧化酶体功能验证实验中发现,PTPRD沉默可导致STAT3信号通路激活,同时过氧化物酶体脂肪酸代谢基因(如ABCD1、ACOX1)显著下调。在模拟代谢性脂肪性肝炎(MASH)的FFA应激模型中,联合沉默STAT3与PTPRD可逆转PTPRD单独沉默引起的基因表达异常,恢复过氧化物酶体功能。这些结果证明,PTPRD与STAT3呈负相关抑制脂肪代谢。
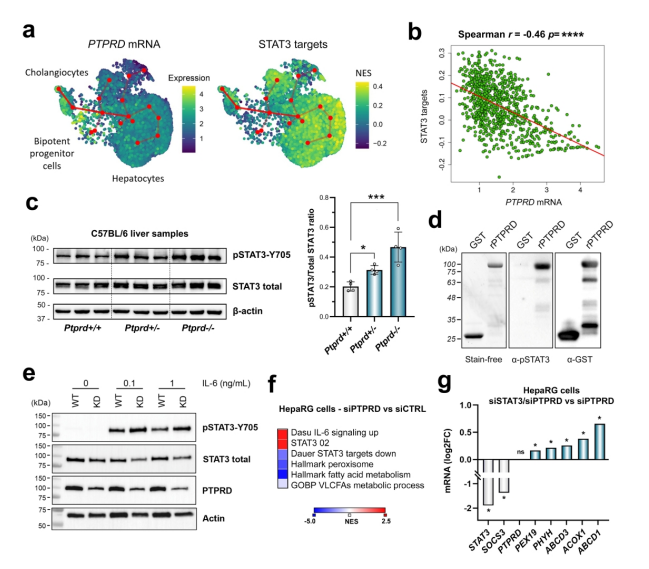
图2:STAT3是肝脏PTPRD底物调控过氧化酶体脂肪酸代谢
a)PTPRD和STAT3靶基因在胆管细胞-肝祖细胞(LPC)-肝细胞的伪时间轨迹(GSE185477)中的表达变化。b)人类肝脏中PTPRD表达与STAT3靶基因表达呈显著负相关(Spearman r=-0.46)。c)通过Western blotting及条带密度定量分析,C57BL/6小鼠中Ptprd基因缺失导致肝脏STAT3 Y705磷酸化(pSTAT3)水平显著升高(Kruskal-Wallis检验),数据以均值±标准差表示。d) pSTAT3是肝细胞中PTPRD的结合伴侣。将5 µg重组GST-PTPRD融合蛋白(rPTPRD)或单独GST蛋白与1 mg IL-6刺激的HepaRG裂解液共孵育,进行免疫共沉淀。同一膜依次进行无染色(总蛋白)、抗pSTAT3(Y705)及抗GST(剥离后)免疫染色。e)基因沉默PTPRD促进IL-6诱导的STAT3磷酸化。利用CRISPR-Cas9(KD)在HepaRG细胞中沉默PTPRD基因,血清饥饿过夜后经IL-6刺激30分钟,通过免疫印迹分析。人源PTPRD抗体检测到胞外域脱落后产生的PTPRD片段。f)PTPRD沉默的HepaRG细胞的基因集富集分析(GSEA)显示STAT3信号通路转录活性增强,脂肪酸代谢相关基因集表达受抑。g)在游离脂肪酸(FFA)处理的HepaRG细胞中,联合沉默PTPRD和STAT3可逆转单独沉默PTPRD引起的基因表达异常,包括过氧化物酶体功能(如PEX19、PHYH)和脂肪酸代谢(如ABCD1、ACOX1)。
注释:ABCD,ATP结合盒亚家族D;ACOX1,酰基辅酶A氧化酶1;FFA,游离脂肪酸;GSEA,基因集富集分析;GST,谷胱甘肽-S-转移酶;PEX19,过氧化物酶体生物合成因子19;PHYH,植烷酰辅酶A 2-羟化酶;PTPRD,蛋白酪氨酸磷酸酶δ;SOCS3,细胞因子信号抑制因子3;STAT3,信号转导及转录激活因子3;VLCFAs,超长链脂肪酸。*p<0.05,***p<0.001,****p<0.0001。
来源:原文图4
PTPRD调控胰岛素抵抗的新途径
本研究进一步发现沉默原代人类肝细胞(PHH)中的PTPRD显著抑制胰岛素诱导的AKT磷酸化(Ser473位点)。通过比较Ptprd杂合与野生型小鼠的肝脏转录组数据,结果显示未折叠蛋白反应(unfolded protein response,UPR)相关基因(MANF、SDF2L1、HSPA5、DNAJB11)在Ptprd缺陷小鼠肝脏与糖尿病信号呈正相关,而与脂代谢调控基因ACACB则呈负相关,所有基因均与胰岛素信号通路呈负相关,并在内质网应激诱导的UPR中被激活,提示PTPRD可能通过抑制UPR调控胰岛素信号,维持代谢稳态。最后,本研究根据肝脏PTPRD表达水平将737名肥胖患者(GSE130991)分为低表达组(后20%)与高表达组(前20%),发现低表达组患者的空腹血糖、糖化血红蛋白(HbA1c)、HOMA2胰岛素抵抗指数、甘油三酯及腰围均显著升高,肝脏UPR基因显著上调,而脂代谢调控因子ACACB表达下调,与Ptprd缺陷小鼠及无CLD个体的研究结果一致。提示肝脏PTPRD通过调控糖脂稳态参与代谢性肝病的发生,其表达异常与临床代谢紊乱表型密切相关。
图3:PTPRD表达受损通过未折叠蛋白反应抑制胰岛素信号
a)在原代人类肝细胞(PHH)中沉默PTPRD后,胰岛素刺激的AKT磷酸化(S473位点)显著减弱(U检验,Western blotting分析,n=4)。b)RNA测序鉴定Ptprd+/-小鼠(n=3)与Ptprd+/+小鼠(n=3)肝脏样本中30个上调基因和29个下调基因(FDR<0.05,Wald检验)。c)Ptprd+/-小鼠差异表达基因的人类同源物与健康个体(n=38,GSE61260)肝脏中PTPRD表达呈显著相关性(Spearman相关性分析)。d)PTPRD相关基因(MANF、SDF2L1、HSPA5、DNAJB11和ACACB)与糖尿病进展及胰岛素信号转录程序的相关性(n=38,GSE61260,Spearman相关性分析)。e)根据肝脏PTPRD表达将肥胖患者分为高表达组(n=147,灰色)和低表达组(n=147,绿色),数据来源于GSE130991。f)肥胖患者临床数据分析显示,低PTPRD表达组空腹血糖、HbA1c、HOMA2胰岛素抵抗指数、血甘油三酯及腰围均显著升高(T检验)。g)低PTPRD表达组患者中MANF和SDF2L1显著上调,ACACB显著下调(T检验)。
注释:ACACB,乙酰辅酶A羧化酶β;AKT,AKT丝氨酸/苏氨酸激酶;DNAJB11,dnaJ热休克蛋白家族(Hsp40)成员B11;HbA1c,糖化血红蛋白;HOMA2,稳态模型评估2;HSPA5,热休克蛋白家族A(Hsp70)成员5;IR,胰岛素抵抗;MANF,中脑星形胶质细胞源性神经营养因子;SDF2L1,基质细胞衍生因子2样1;PTPRD,蛋白酪氨酸磷酸酶δ。*p<0.05,**p<0.01,***p<0.001,****p<0.0001。
来源:原文图5
临床转化价值与未来方向
综上所述,本研究揭示了PTPRD可通过调节STAT3信号通路和过氧化物酶体脂肪酸代谢,影响肝脏的代谢功能。此外,PTPRD的缺失还可能通过UPR影响胰岛素信号通路,导致胰岛素抵抗。这些发现提示肝脏PTPRD表达水平可作为代谢性肝病风险的潜在生物标志物,并为开发针对PTPRD的治疗策略提供了理论基础,可能有助于改善代谢性肝病患者的临床预后。此外,研究者强调需进一步验证PTPRD在血液中的可检测性及其与肝病分期的相关性。
引证本文
Armando Andres Roca Suarez, Frank Jühling, Julien Moehlin, Laurent Mailly, Alessia Virzì, Nicolas Brignon, Sarah C Durand, Marine A Oudot, Eugenie Schaeffer, Romain Martin, Laura Meiss-Heydmann, Charlotte Bach, Zakaria Boulahtouf, Lea Girard, Emma Osswald, Carole Jamey, Daniel Brumaru, Nassim Dali-Youcef, Atish Mukherji, Maria Saez-Palma, Barbara Testoni, Fabien Zoulim, Bhuvaneswari Koneru, Naoto Fujiwara, Yujin Hoshida, Emanuele Felli, Patrick Pessaux, Michel L Tremblay, Romain Parent, Catherine Schuster, Thomas F Baumert, Joachim Lupberger - Protein tyrosine phosphatase delta is a STAT3-phosphatase and suppressor of metabolic liver disease: eGastroenterology 2025;3:e100159.
https://doi.org/10.1136/egastro-2024-100159

