罕见病专栏|肢体乏力10余年——远端型遗传性运动神经病
时间:2025-02-28 12:25:15 热度:37.1℃ 作者:网络
摘 要 远端型遗传性运动神经病(distal hereditary motor neuropathy, dHMN)是罕见的遗传性周围神经病,由于其发病率低,临床表现异质性强,临床易误诊或漏诊。本文报告1例可疑远端型遗传性运动神经病。患者发病十年,以隐匿性肢体无力起病,伴有肌肉萎缩,无感觉异常,神经科查体及神经电生理检查均提示为远端对称性运动纤维轴索损害,详细分析患者临床资料及神经电生理检查特点,符合远端型遗传性运动神经病的特点,基因筛查提示患者有BSCL2基因突变。经营养神经等对症治疗,随访将近1年,患者临床症状及电生理检查无明显进展。该病例提示临床工作中应注意遗传性周围神经病的可能,善于分析临床及电生理检查资料,必要时采用基因诊断,做到早发现、早确诊。
关键词
远端型遗传性运动神经病;BSCL2;肌电图
远端型遗传性运动神经病(distal hereditary motor neuropathy, dHMN)即远端脊髓性肌萎缩症,是少见的遗传性周围神经病,典型临床表现为长度依赖性的运动无力和肌肉萎缩,通常没有感觉症状或者感觉症状轻微。该病起病隐匿,容易误诊或漏诊。本文报告我院门诊2021年诊治1例可疑dHMN患者,以增加临床医生对dHMN的认知。
1 临床资料
患者,男,40岁,主因“四肢乏力10余年,加重两年”于2021年11月就诊我院。患者于2008年无明显诱因出现肢体麻木,否认当时有肢体乏力及肌肉萎缩,于外院就诊,当时曾行肌电图检查,提示周围神经损害(具体不详)。后肢体麻木情况自行缓解,未再诊治。2010年逐渐出现双下肢乏力,表现为穿拖鞋走路容易掉鞋,后逐渐加重,发展为不能穿拖鞋走路。2013年前后出现双手无力,开始抓东西费力、不灵活,尤其用刀切东西时明显。后肢体乏力情况逐渐加重,并出现手部肌肉萎缩。就诊时穿运动鞋尚可行走,手仍可以提东西。患者无明显肢体麻木及疼痛。发病以来食欲尚可,睡眠欠佳,易心烦、紧张,易担心、害怕,大小便正常,体质量无明显变化。
既往史:否认糖尿病等其他特殊疾病,自幼按计划接种疫苗,否认特殊药物及毒物接触史,否认吸烟及酗酒史。
体格检查:体型消瘦,跨阈步态。心、胸、腹部查体未见异常。神经系统查体:高级皮层功能未查及明显异常,双侧咽反射稍减弱,余脑神经未查及异常。双手大小鱼际肌及骨间肌萎缩(图1),双足肌肉萎缩,双侧小腿肌肉轻度萎缩(图2),大腿及双前臂、上臂肌肉无明显萎缩。四肢近端肌力4级,双手握力3级,双足背屈不能(0级),双侧高弓足,无明显感觉障碍,四肢腱反射(-),病理征阴性。
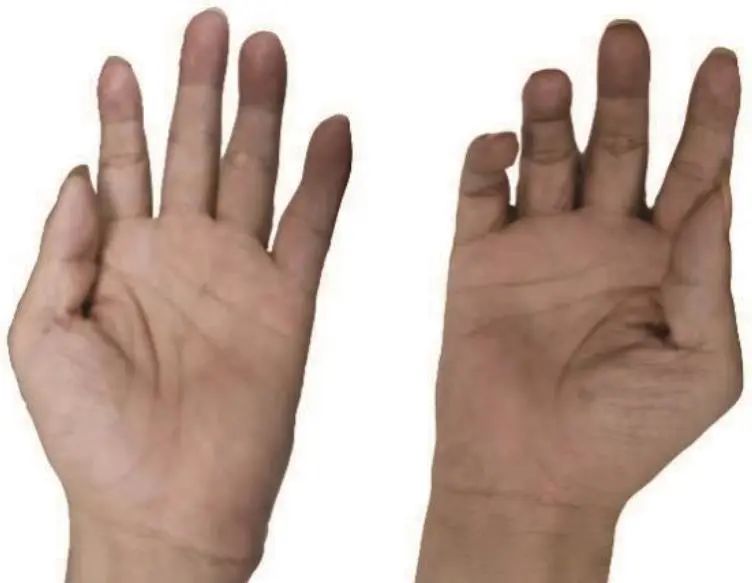
图1 患者双手
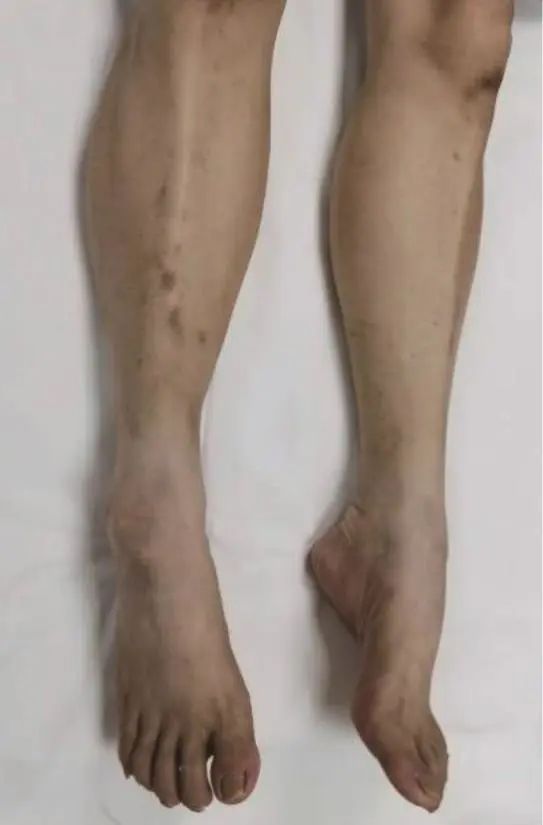
图2 患者双足及小腿
肌电图感觉神经传导:左右正中神经、左右尺神经、左右腓浅神经、左右腓肠神经感觉传导速度及波幅正常范围。运动神经传导:左右正中神经、左侧尺神经运动传导末端潜伏期正常范围,波幅低,肘-腕传导速度稍慢;右侧尺神经、左右腓总神经、胫神经运动传导末端潜伏期及传导速度正常范围,波幅明显降低。F波及H反射:左右正中神经、左右尺神经F波出现率低,潜伏期正常范围,左右胫神经F波出现率及潜伏期正常范围;左右胫神经H反射未引出。针电极肌电图:右侧小指展肌见自发电位,轻收缩仅获得单个运动单位电位,波幅低,时限偏短,右侧胫骨前肌放松状态下见自发电位,轻收缩未能获得运动单位电位,右侧肱二头肌、右侧斜方肌、右侧股四头肌放松状态下未见自发电位,轻收缩部分运动单位电位时限延长。T10脊旁肌未见自发电位。
患者DNA标本经高精度外显子测序检测到患者BSCL2基因存在杂合突变c.974 dup(p.I326Hfs*12),见图3。随后对患者的儿子和女儿进行了突变位点的检测,其儿子也发现了BSCL2基因该位点的突变。但其儿子目前尚无不适。
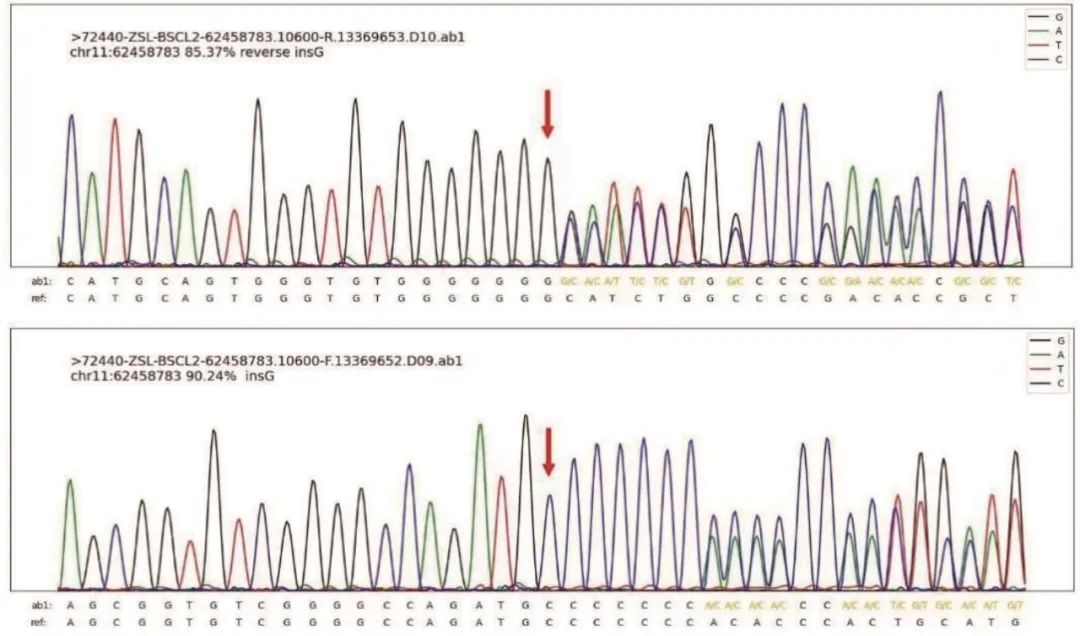
图3 患者测序结果
诊断治疗及随访:本例患者表现为远端型运动受累的下运动神经损害,主要是双足、小腿和手部的肌肉萎缩及无力,左右基本对称,没有感觉障碍。电生理检查也证实患者为周围神经损害,感觉纤维正常,仅有运动纤维受损,为远端对称性轴索损害,多节段神经传导未见传导阻滞。针电极肌电图检查提示远端神经源性损害较近端明显,因而符合周围神经的分布的特点。结合患者病史比较长,进展缓慢,没有上运动神经元损害的体征,我们考虑遗传性运动神经病可能。给予甲钴胺、维生素B1及维生素B6等对症治疗,2022年7月随访患者症状无明显进展,肌电图检查较前无明显变化。
2 讨论
本例患者为慢性起病,缓慢进展,以远端肌肉萎缩无力为主,左右基本对称,没有感觉纤维受损和上运动神经损害的表现和体征,符合下运动神经损害的临床表现,神经传导检查提示仅周围神经运动纤维受损,定位基本明确为下运动神经。针电极肌电图提示患者神经源性损害远端重于近端,符合周围神经损害的特点,因而患者定位诊断为:周围神经、运动纤维。患者既往病史和辅助检查结果不支持中毒、代谢及肿瘤因素导致的周围神经损害,结合慢性起病,缓慢进展,主要考虑遗传或免疫的可能性较大。但是患者拒绝进一步的腰椎穿刺术及周围神经抗体检查,因而未能完全排除免疫因素。但患者病史有十余年,随访近1年期间症状和体征均无明显变化,电生理检查均提示纯运动纤维受累,周围神经损害具有对称性,未发现传导阻滞及波形离散,不符合多灶性运动神经病和慢性炎症性脱髓鞘性多发神经根神经病等免疫性周围神经损害的特点。且患者有明显的弓形足,因而考虑患者为遗传性周围神经病可能,且为纯运动纤维受损类型。这些特点符合远端型遗传性运动神经病的临床特点。远端型遗传性运动神经病是由下运动神经元功能障碍引起的一组临床和遗传异质性疾病,多数患者在青少年期发病,但也有部分患者(如本例)在成人期发病[1]。dHMN 典型临床表现是长度依赖性的运动无力和肌肉萎缩,多数会先影响足部肌肉,逐渐累及小腿和手部肌肉,部分患者可以先出现手部症状,通常没有感觉症状或者感觉症状轻微[2]。而遗传性周围神经病最常见的类型——腓骨肌萎缩症(Charcot-Marie-Tooth,CMT)亦称为遗传性运动感觉神经病,通常伴有感觉症状,神经传导检查可以发现感觉纤维明显受损,本例患者临床表现没有感觉症状,电生理检查也进一步排除了临床下的感觉纤维受损,因而最后的诊断我们考虑为单纯运动纤维受累的远端型遗传性运动神经病。
神经电生理检查是周围神经病的重要辅助检查之一。dHMN神经电生理多表现为运动轴索损害,部分患者可以有轻度的传导速度减慢[3-4]。本例患者神经传导检查结果提示患者周围神经运动纤维轴索受损为主,多节段检查未发现传导阻滞。针电极肌电图检查提示远端损害较近端损害明显,呈周围神经分布特点,符合dHMN的电生理表现。下肢神经传导腓总神经运动波幅降低较胫神经更为明显,这与既往报道[5]也一致。
目前已报道约30种dHMN致病基因,这些基因编码的蛋白质参与细胞内各种生化活动,如蛋白折叠(HSP1,HSP8,DNAJB2)、RNA代谢(IGHMBP2,SETX,GARS,AARS)、轴浆运输(BICD2,DYNC1p,DCTN1)和阳离子通道功能(ATP7A,TRPV4,KCC3)等,但仍有超过一半的dHMN先证者致病基因不明[6]。传统的一代测序基因检测阳性率仅为15%~20%[7-8]。随着二代测序技术的发展,全外显子组测序技术提高了基因诊断的阳性率,更多新的基因突变被发现。BANSAGI B等对64例dHMN先证者进行二代测序,发现基因诊断的阳性率增至30%~40%[6]。
本例患者基因检测发现BSCL2突变是远端型遗传性运动神经病的最常见突变基因之一, dHMN患者7%~12%存在BSCL2突变[9-11]。BSCL2 编码的Seipin蛋白是一种主要位于内质网的跨膜蛋白。它通过调节AMPA受体水平来调节突触传递[12] 。2001年MAGRE等[12]首次确定了BSCL2为先天性全身性脂肪营养不良2型的候选基因。目前BSCL2被确定为远端型遗传性运动神经病的突变点位包括p.N88S和p.S90L[13-14]。本例患者BSCL2为c.974 dup(p.I326Hfs*12)突变,根据美国 ACMG 变异分类指南(PMID: 25741868, 31690835),这个变异为2 类,即可能致病。我国近期一项关于先天性全身性脂肪营养不良的研究也发现BSCL2基因c.974 dup突变为该疾病常见的突变点之一[15],这证明该位点的突变是具有致病性的。但目前尚未查到此突变与遗传性运动神经病的相关报告。患者儿子也携带了相同的突变,但目前尚未达到发病年龄,也无临床表现,需要进一步随访,才能明确。因此BSCL2的c.974 dup(p.I326Hfs*12)是否为本患者的致病突变尚无定论。
3 点评
既往认为遗传性疾病多为儿童期或者青少年起病,中年起病较少。随着二代测序技术的发展,其临床应用越来越普遍,许多既往未能查明原因的周围神经神经病,经过测序发现了基因异常,从而明确诊断,并因此发现了许多中年发病的遗传性周围神经病。虽然发现的BSCL2基因突变位点是否本患者的致病基因还需要进一步的随访观察,但综合患者的临床资料,我们认为患者符合远端型遗传性运动神经病的诊断,同时我们也会随访患者儿子的情况,必要时行肌电图检查,寻找临床下周围神经损害的依据。
参考文献:
1. ROSSOR A M, KALMAR B, GREENSMITH L, et al. The distal hereditary motor neuropathies[J]. J Neurol Neurosurg Psychiatry, 2012, 83: 6-14.
2. FRASQUET M, ROJAS-GARCÍA R, ARGENTE-ESCRIG H, et al. Distal hereditary motor neuropathies: mutation spectrum and genotype-phenotype correlation[J]. Eur J Neurol, 2021, 28(4): 1334-1343.
3. AUER-GRUMBACH M, SCHLOTTER-WEIGEL B, LOCHMULLER€ H, et al. Phenotypes of the N88S Berardinelli-Seip congenital lipodystrophy 2 mutation[J]. Ann Neurol, 2005, 57: 415-424.
4. BANSAGI B, GRIFFIN H, WHITTAKER R G, et al. Genetic heterogeneity of motor neuropathies[J]. Neurology, 2017, 88(13): 1226-1234
5. 周美鸿, 朱敏, 洪道俊. 中年女性, 右下肢无力5个月-远端型遗传性运动神经病[J]. 中国神经精神疾病杂志, 2020, 46(6): 380-384.
6. DIERICK I, BAETS J, IROBI J, et al. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study[J]. Brain, 2008, 131(Pt 5): 1217-1227.
7. ROSSOR A M, EVANS M R, REILLY M M. A practical approach to the genetic neuropathies[J]. Pract Neurol, 2015, 15(3): 187-198.
8. YOLAINE O, ARMELLE M, PHILIPPE L, et al. Clinical and electrophysiological features in a French family presenting with seipinopathy[J]. Neuromuscul Disord, 2015 , 25(2): 161-164.
9. DIERICK I, BAETS J, IROBI J, et al. Relative contribution of mutations in genes for autosomal dominant distal hereditary motor neuropathies: a genotype-phenotype correlation study[J]. Brain, 2008, 131(Pt 5): 1217-1227.
10. ROHKAMM B, REILLY M M, LOCHMÜLLER H, et al. Further evidence for genetic heterogeneity of distal HMN type V, CMT2 with predominant hand involvement and Silver syndrome[J]. J Neurol Sci, 2007, 263(1-2): 100-106.
11. WEI S, SOH SL, QIU W, et al. Seipin regulates excitatory synaptic transmission in cortical neurons[J]. J Neurochem, 2013, 124(4): 478-489.
12. MAGRE J, DELEPINE M, KHALLOUF E, et al. Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13[J]. Nat Genet, 2001, 28: 365-370.
13. WINDPASSINGER C, AUER-GRUMBACH M, IROBI J, et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome[J]. Nat Genet, 2004, 36: 271-276.
14. IROBI J, VAN DEN BERGH P, MERLINI L, et al. The phenotype of motor neu-ropathies associated with BSCL2 mutations is broader than Silver syndrome and distal HMN type V[J]. Brain, 2004, 127: 2124-2130.
15. SU X, LIN Y, LIU L, et al. Features of BSCL2 related congenital generalized lipodystrophy in China: long-term follow-up of three patients and literature review.J Pediatr Endocr Met. 2023, 36(1): 74-80.
【引用格式】谢春格,陈洁玲,甘蓉,等. 肢体乏力10余年——远端型遗传性运动神经病[J]. 中国神经精神疾病杂志,2023,49(5):318-320.
【Cite this article】XIE C G,CHEN J L,GAN R,et al. Limb weakness for 10 years - Distal inheritary motor neuropathy?[J]. Chin J Nervous Mental Dis,2023,49(5):318-320.
DOI:10.3969/j.issn.1002-0152.2023.05.012

