“有温度的检验人”——一例FIB危急值的踏“血”寻踪
时间:2023-01-08 18:00:46 热度:37.1℃ 作者:网络
1
前 言
遗传性异常纤维蛋白原血症是一种由常染体基因突变引起的染色体显性遗传性疾病;Philippe de Moerloose报道遗传性纤维蛋白原缺陷症发病率约1/106,属于罕见遗传性疾病[1,2]。纤维蛋白原是一种肝细胞分泌的相对分子量340kD的可溶性纤维蛋白前体;由位于4号常染色体长臂Q23-Q32区的FGA、FGB和FGG基因编码[3,4]。
异常纤维蛋白原血症的临床表现呈多样性,主要可以概括为:(1)无症状:大多数患者无临床表现,在术前筛查时才确诊。(2)出血:部分患者(约20%)会出现出血症状,多为轻度出血。(3)血栓:部分患者(约占20%)会有下肢静脉血栓的风险。(4)少数患者既有出血表现又有血栓形成的表现。
2
案例经过
患者,女性,58岁,藏族,已婚,农民,青海籍,2021年11月以“宫颈癌术后放化疗后3年,远处转移1年余化疗后”为主诉,门诊以“宫颈中-低分化鳞状细胞癌IIIC2p期术后化疗后”收住入院。患者于3年前因“阴道淋漓出血”就诊外院,明确诊断为鳞状细胞癌。
于2019年1月30日行“经腹广泛子宫全切术+双侧附件切除术+盆腔淋巴清扫术+腹主动脉旁淋巴清扫术+腹腔引流术”,2019年3月5日行术后辅助放化疗,DP方案。此次来我院住院复查准备再次化疗。父母已逝,病因不详,否认三代以内近亲婚育史。
凝血报告纤维蛋白原(fibrinogen,FIB)结果为0.70g/L(如下图1),触发科室危急值。在排除标、仪器、试剂等会影响检验结果的其他因素后,再次复测标本,确认检测结果无误,查看其他同期患者检验结果,未发现会影响FIB急剧下降的异常原因。
查看凝血酶原时间(prothrombin time,PT)演算法所测得的FIB结果是3.1g/L,Clauss法结果FIB的活性检测与衍算法检测比值小于0.7,考虑该患者是异常纤维蛋白原血症,通过与临床沟通建议患者亲属进一步检测FIB,并加做基因检测,最终证实为遗传性家族性异常纤维蛋白原血症。

图1
3
案例分析
临床角度:
近年来,随着检验科与临床的主动沟通,临床医生越来越愿意与检验人员探讨有关检测项目和异常结果的问题。临床将纤维蛋白原缺陷症大体分为两类:一类FIB数量异常,包括低纤维蛋白原血症和无纤维蛋白原血症;第二类为FIB质量异常,包括异常纤维蛋白原血症和低异常纤维蛋白原血症。
异常性纤维蛋白原血症是FIB基因缺陷导致FIB分子结构异常与功能缺陷的一种遗传性疾病,绝大多数为常染色体显性遗传。
异常纤维蛋白原血症的临床表现呈多样性,主要可以概括为:(1)无症状:大多数患者无临床表现,在术前筛查时才确诊。(2)出血:部分患者(约20%)会出现出血症状,多为轻度出血。(3)血栓:部分患者(约占20%)会有下肢静脉血栓的风险。(4)少数患者既有出血表现又有血栓形成的表现。
治疗:对于无症状的患者无需治疗。有出血表现或需要手术者常采用替代疗法,补充冷沉淀、纤维蛋白原制剂及血浆等。有血栓形成倾向的患者需采用抗凝治疗,如肝素、华法林、阿司匹林及新型口服抗凝药物等。
疾病发展和转归:对于大部分无症状的患者,本病不需治疗。对于患者的生活质量,生育及预期寿命等基本无影响,预后好。若不接受正规治疗,部分出血严重患者,可出现贫血表现,影响患者的生活质量,但极少数患者的出血会危及生命。以血栓形成为主要表现的患者,可能由于下肢深静脉血栓形成导致肺栓塞引起死亡,动脉栓塞时,可能导致相应部位坏死,如下肢动脉栓塞导致截肢,冠状动脉栓塞导致心肌梗死或心肌缺血等。
经过正规治疗后,以出血为主要表现的患者,基本不影响患者的生活质量及预期寿命。对于血栓形成的患者,需长期服用抗凝药物,并防止出血,若无其他严重并发症,对生活质量及寿命影响较小。本病无法治愈,有症状的患者需要终身治疗。
4
检验角度:
究竟何原因导致患者FIB如此低呢?我们开始逐一进行排查:
1.排除仪器和试剂的系统问题;
2.排除标本采集的问题;
3.排除药物干扰的问题:某些药物如蛇毒血凝酶等也可能导致FIB检测结果偏低,通过和临床主管医生沟通排除治疗和药物干扰的因素,临床医生考虑是疾病本身所致。
查看凝血酶原时间(prothrombin time,PT)演算法所测得的FIB结果是3.1g/L,Clauss法结果FIB的活性检测与衍算法检测比值小于0.7,再次询问患者相关的病史和家族史,并告知主管医生此患者有可能是一例异常纤维蛋白原血症的患者,建议医生告知患者直系亲属来检测FIB,怀疑是家族性遗传性的异常纤维蛋白原血症。
主管医生按照我们的建议告知患者让其女儿前来检查(父母已逝,妹妹已失去联系)。随后一段时间曾两次和临床主管医生联系其患者女儿前来做检测,第二次医生告知我患者女儿在某县级医院检测过了,结果正常,我挂完电话后仍然不甘心,又再次拨通电话告诉医生对患者说她女儿来我院检验科,免费为其抽血检测。终于一个月后患者女儿来我科检测,Clauss法检测结果如下图2,PT演算法结果为3.3g/L。

图2
结果与最初预料基本一致,第一时间告知主管医生其患者女儿的FIB检测结果,至此基本可以初步判定这是家族性遗传性异常纤维蛋白原血症。然而事情到这里还没有结束,患者女儿36岁,追问患者女儿生育史。患者女儿现育有两子,继续追问既往史,其否认分娩时发生过血栓或出血事件。
于是我告诉患者女儿,等寒假时带两个儿子来我院检查,仍然免费给两个孩子抽血检测。又隔了一个多月,2022年1月7日,患者女儿带两个儿子(分别是8岁和10岁)来找我(图3),我询问两个孩子是否有特殊临床表现和体征,除了10岁孩子手掌经常出现如下红斑以外(图4),没有特殊临床表现和阳性体征,两个孩子的Clauss法检测结果如下图5-6,8岁孩子和10岁孩子PT演算法检测结果分别为3.2g/L和3.1g/L。

图3

图4

图5
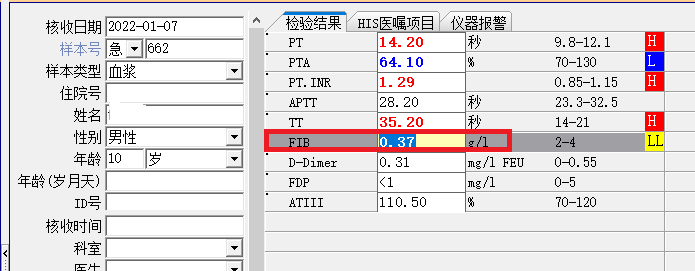
图6
看到以上检测结果后马上和主管医生进行沟通,高度怀疑是“家族性遗传性异常纤维蛋白原血症”,建议患者女儿进一步基因检测,以明确诊断。我联系到广州知力医学诊断技术有限公司医学总监-赖建禧教授,有幸赖教授正在做全国异常纤维蛋白原血症基因的课题,承诺以优惠价帮助患者检测基因。因考虑经济问题,只做患者女儿的基因,检测结果如下图7。

图7
基因检测结果为FGA杂合突变,突变位点为FGA:NM-000508:exon2:cG104A:p.R35H。经华中科技大同济医学院附属同济医院血栓与止血实验室负责人唐宁教授将此基因结果与纤维蛋白原基因突变库中的突变位点核对后,提示无血栓及出血风险,我将此核对结果及时反馈予主管医生、患者及其女儿。
作为新时代的检验人,我们早已不再是检验“匠人”了,虽然检测出来的是数字,但关注的是数字背后的“真相”。当检测出异常结果时,我们绝不轻易发出,我们会按照科室流程一一排查;会去分析产生异常结果的原因;会翻阅病历,查看病程记录;会核对遗嘱用药,查阅资料;会去临床沟通、去患者床边问诊查看,会建议患者亲属前来检查,为了让其亲属愿意来检测,有时我们甚至免费。
同时我们会请教专家,通过网络专业群进行讨论。会和临床主动沟通,有时为了寻找一个无法解释的异常检验结果的原因真是废寝忘食好几天、几个月甚至更长一段时间,总想查个清楚,给临床提供一些诊疗线索和帮助。
我们实验室每台设备都已设置了FIB的PT演算法和Clauss法,按照报道,当PT演算法结果/Clauss法结果>1.43,或Clauss法结果/PT演算法结果<0.7时,可提示异常纤维蛋白原血症的可能,目前纤维蛋白原的检测,还有抗原检测,主要包括ELISA、比浊法、沉淀法等。基因型检测:FIB由3对肽链构成,可以通过检测FGA、FGB、FGG基因来明确是否有突变,是目前确诊异常性纤维蛋白原血症的最佳方法。质谱分析通过飞行时间换算出小肽段的质量,经计算机软件构建蛋白质一级结构信息,获得FIB肽链氨基酸序列的特征图谱。
5
知识拓展
遗传性异常纤维蛋白原血症是一种由常染体基因突变引起的染色体显性遗传性疾病;Philippe de Moerloose报道遗传性纤维蛋白原缺陷症发病率约1/106,属于罕见遗传性疾病[1,2]。纤维蛋白原是一种肝细胞分泌的相对分子量340kD的可溶性纤维蛋白前体;由位于4号常染色体长臂Q23-Q32区的FGA、FGB和FGG基因编码[3,4]。
正常情况下,肝脏组织中转录FIB编码基因,由核糖体翻译合成三条同源多肽链:610个氨基酸残基的Aα链、461个氨基酸残基的Bβ链和411个氨基酸残基的γ链;转移到粗面内质网的多肽链加工连接形成Aα-γ、Bβ-γ中间体,进一步结合为Aα-Bβ-γ三聚体亚基结构;二硫键将两个Aα-Bβ-γ三聚体亚基聚合为(Aα-Bβ-γ)2六聚体[5,6],FIB作为血浆中含量最高的凝血因子I,主要参与血栓形成、纤维蛋白溶解、血小板聚集等,参与凝血、止血、伤口修复、血管新生、肿瘤生长与转移过程[7]。
纤维蛋白原的缺乏包括获得性和遗传性两种,其中遗传性纤维蛋白原缺乏症又分为I型FIB减少的无纤维蛋白原血症和低纤维蛋白原血症、II型FIB质量异常的异常纤维蛋白原血症(dysfibrinogenemia,DYS)和低异常纤维蛋白原血症(hypodysfibrinogene-mia,HYPODYS)[8,9]。
遗传性异常纤维蛋白原血症常见的遗传性基因突变类型有片段缺失、启动子突变、剪接点突变、框移突变、无义突变和错义突变等。FIB多表现为FGA编码区的Aα链突变、FGB编码区的Bβ链突变和FGG编码区的γ链突变;致使转录mRNA密码子异常,多肽链氨基酸顺序、种类改变,表达蛋白质一级肽链氨基酸序列异常,功能蛋白分子空间结构变异、蛋白功能单位变化,主要表现为纤维蛋白肽释放障碍、纤维蛋白单体聚合障碍或XIIa介导的交联障碍等[10],即纤维蛋白原功能改变、数量减少、质量下降等。
临床表现为血浆中纤维蛋白原浓度明显减少甚至缺如;纤维蛋白原完全缺乏程度为遗传性无纤维蛋白原血症[11](Congential afibrinogenemia);而循环血液中纤维蛋白原水平可以测到,但又低于1.5 g/L时称为遗传性低纤维蛋白原血症(Congential hypofibrinogenemia);若循环血中纤维蛋白原水平正常但功能异常的情况下定义为遗传性异常纤维蛋白原血症(Congential dysfibrinogenemia,CD)[12]。
该病的临床症状[13]表现为血栓和/或出血倾向,且因纤维蛋白原缺乏程度不同而表现出不同的症状,缺乏最严重的遗传性无纤维蛋白原血症多呈现出血症状,部分统计示约85%左右患者呈脐带出血;72%患者表现为血肿、鼻出血、月经量多、牙龈出血等;54%的关节出血等。遗传性低纤维蛋白原血症患者一般不表现出临床症状,但机体损伤、血管破裂等创伤时,机体呈现明显的出血倾向。
异常纤维蛋白原血症患者通常无临床症状,仅1/4患者有出血表现,其中1/5的患者在青年时期出现下肢深静脉血栓、血栓性静脉炎、肺栓塞等血栓事件。Haverkate等[7]研究显示,约55%的DYS患者无任何临床症状,25%的患者有出血史,20%的患者有血栓倾向且主要是静脉血栓。出血多见于纤维蛋白肽释放障碍及FIB交联缺陷,而血栓栓塞多见于纤维蛋白单体聚合障碍。同时DYS血栓形成的机制:(1)异常的FIB与凝血酶结合缺陷,从而导致凝血酶水平升高;(2)异常FIB形成的纤维蛋白凝块抗纤溶酶降解[8-9]。
CD患者因Fg减少或缺失而降低Fg作用,同时CD患者本身有出血和或血栓风险,表现在沟通母体与胎儿的胎盘中则引起胎盘早剥、自发性流产、早产等;产后大出血、血栓栓塞等高危风险[14,15]。故而确诊患者在妊娠期及分娩前后动态检测凝血功能,维持血浆中Fg水平。
参考2016年专家共识[16],维持妊娠前6个月血浆中Fg/C水平波动于0.5-1.0 g/L,妊娠后3个月及围生期维持于1.0-2.0 g/L;需注意在分娩过程中保持在1.5 g/L以上。妊娠分娩间可采用输注浓缩Fg达到抗凝治疗及预防血栓目的。同时女性生理期子宫周期性出血,CD患者因出血倾向引起月经过多、月经期延长;或血栓可能引起月经期间腹痛加重等。
男性患者虽然可避免生理性出血、血栓风险,但有明确研究报道了CD患者深静脉血栓发病率显着增高;且于不同年龄段有较高的静脉血栓、动脉血栓及大出血风险[17,18]。故而CD患者家系中男性、女性患者均需要进行健康宣教、长期随访,预防创伤、出血风险、血栓发生。
6
案例总结
通过研究FGA突变致遗传性异常纤维蛋白原血症患者临床症状及诊治方案,并分析家族发病情况,明确CD的基因突变位点、探讨发病机制,以提升临床医生对CD的认识、诊断、治疗,为优生优育和产前基因诊断提供理论依据。做一个“有温度的检验人”,在实际检验工作中,我们要始终秉承精益求精的探索精神,勇于探索的创新思维,当发现异常结果时善于与临床沟通并仔细分析,逐一排除影响因素,我们眼里不仅看到检验结果,还要关注到检验结果后面的患者,尽最大努力为临床医生提供有利于患者诊断、治疗和预后的检验信息,用实际行动诠释“为临床及患者优质服务”的内涵。
7
专家点评
华中科技大同济医学院附属同济医院 唐宁教授
遗传异常纤维蛋白原血症患者可表现为无症状、出血或血栓,在管理策略上与其他原发/继发因素所致纤维蛋白原缺乏也有区别,因此遗传性异纤的准确、及时诊断对于患者处置非常重要。本文作者从实验室常规检查出发,根据检验路径主动建议下一步检查,并积极随访患者,帮助患者及时确诊,避免了不必要的诊疗,体现了检验科医/技师的临床咨询能力。
参考文献
[1]骆娟,段苏容,王华.1例遗传性异常纤维蛋白原血症的家系分析和诊断报告[J].四川大学学报(医学版),2022,53(01):171-174.
[2]郁婷婷,傅启华.罕见病诊治思考与展望[J].检验医学,2021,36(02):119-121.
[3]余曼.遗传性纤维蛋白原缺陷症患者的纤维蛋白原基因突变鉴定和特征分析研究[D].南昌大学,2021.DOI:10.27232/d.cnki.gnchu.2021.000553.
[4] Zhou J,Ding Q,Chen Y,et al.Clinical features and molecular basis of 102 Chinese patients with congenital dysfibrinogenemia [J].Blood cells,molecules diseases.2015,55(4):308-315.
[5]王甜甜,邵静茹,王杰,等.新型FGG基因突变导致遗传性纤维蛋白原缺陷症的研究.中国实验血液学杂志,2021,29(2): 586–590.
[6]李丹丹.两例遗传性异常纤维蛋白原血症的临床和分子发病机制研究[D].河北医科大学,2019.
[7]周景艺.遗传性异常纤维蛋白原血症的临床和分子发病机制研究[D].上海交通大学, 2016.DOI:10.27307/d.cnki.gsjtu.2016.003213.
[8]LEBRETON A,CASINI A.Dignosis of congenital fibrmogen disomdlers[J]Ann Biol CLin(Paris),2016,74(4):405-4I2.
[9]宋景春.重症患者纤维蛋白原缺乏症的现代诊疗观点[J].东南国防医,2018,20(05):454-458.
[10]黄丹丹,蔡挺,张顺,黄左安,郭莳雨,丁秋兰,戴菁,王学锋.1例遗传性异常纤维蛋白原血症的鉴定及分子发病机制研究[J].临床检验杂志,2019,37(09):675-679.DOI:10.13602/j.cnki.jcls.2019.09.08.
[11]Zhang Y, Zuo X, Teng Y. Women With Congenital Hypofibrinogenemia / Afibrinogenemia : From Birth to Death. Clin Appl Thromb Hemost. 2020 Jan-Dec;26:1076029620912819.
[12] Zhou J, Xin Y, Ding Q,et al. Thromboelastography predicts risks of obstetric complication occurrence in (hypo)dysfibrinogenemia patients under non-pregnant state. Clin Exp Pharmacol Physiol. 2016 Feb;43(2):149-56.
[13]周伟杰,闫婕,邓东红,林发全.遗传性异常纤维蛋白原血症的诊断[J].中华检验医学杂志,2020(04):406-407-408-409-410.
[14]苏日娜,杨慧霞.遗传性纤维蛋白原异常合并妊娠的研究进展[J].中华妇产科杂志,2018,53(01):62-64.
[15]罗莎莎,杨丽红,谢海啸,等. 1例遗传性异常纤维蛋白原血症导致胎停育.临床检验杂志,2020,38(3): 187–190.
[16]Casini A, de Moerloose P, Neerman-Arbez M. Clinical Features and Management of Congenital Fibrinogen Deficiencies. Semin Thromb Hemost. 2016 Jun;42(4):366-74.
[17]Casini A, Blondon M, Lebreton A, et al. Natural history of patients with congenital dysfibrinogenemia[J]. Blood. 2015;125(3):553-61.
[18]de Moerloose P, Boehlen F, Neerman-Arbez M. Fibrinogen and the risk of thrombosis. Semin Thromb Hemost. 2010 Feb;36(1):7-17.

