AJHG发文:人类精子发生过程中的全基因组DNA甲基化变化与男性不育有关
时间:2024-06-04 21:02:11 热度:37.1℃ 作者:网络
越来越多的证据表明,男性生殖细胞甲基化组的建立并不局限于胚胎发育时期,而是精子发生过程中持续到成年期。特别是在减数分裂的早期阶段,当复制和重组发生时,基因组是低甲基化的。但人们对于人类精子发生过程中的DNA甲基化,以及该过程异常是否影响精子发生和功能的了解仍有限。
表观遗传重塑(包括甲基化组的重编程)对于哺乳动物的细胞命运决定至关重要。已有研究表明,参与DNA甲基化机制的酶功能受损会导致精子发生的一系列障碍,因此科研人员推测异常DNA甲基化可能会影响生殖细胞。在减数分裂期间,基因组在很大程度上依赖于转座元件(TEs)的抑制,主要通过表观遗传机制(包括DNA甲基化等)来实现,这提示抑制TE对于维持胚系基因组完整性至关重要。但在精子发生过程中,生殖细胞中不同TE的DNA甲基化保护作用及其与男性不育的关系,特别是在减数分裂的低甲基化阶段,仍有待研究。
近日,德国明斯特大学科研人员在The American Journal of Human Genetics 发表了题为“Genome-wide DNA methylation changes in human spermatogenesis”的文章,通过对人类男性生殖细胞进行全基因组甲基化分析,发现人类精子发生与甲基化组的表观遗传重塑有关。在初级精母细胞中的DNA甲基化整体下降后,精母细胞/精子的特定区域会出现选择性的再甲基化,这表明初级精母细胞基因组的低甲基化不仅仅是DNA复制的短暂副作用,也是建立精母细胞/精子特异性甲基化组所必需的。
此外,研究团队还发现不育男性生殖细胞的DNA甲基化存在显著差异,特别是在基因间区和TE,并证明了在精子发生过程中不同的TE会发生不同程度的重编程。总之,该研究揭示了DNA甲基化变化在人类精子发生过程中的重要作用,并指出DNA甲基化改变与精子发生失败之间存在关联。

文章发表在AJHG
主要研究内容
初级精母细胞全基因组DNA甲基化水平降低
为研究人类精子发生过程中全基因组DNA甲基化的变化,研究团队对来自三名精子发生正常男性(CTR)的生殖细胞进行全基因组甲基化测序(EM-seq),包括未分化、分化的精原细胞,初级精母细胞以及精细胞/精子,平均每个样本捕获28,049,115个CpG位点。分析结果显示,未分化、分化精原细胞和精细胞/精子的DNA甲基化平均水平相对较高。有趣的是,与其他生殖细胞类型相比,初级精母细胞的总体DNA甲基化水平显著较低。
为阐明初级精母细胞中的DNA低甲基化是否为随机发生,研究团队分析了基因体以及转录起始位点(TSS)和转录结束位点(TES)上下游5000bp的DNA甲基化。结果显示,初级精母细胞DNA甲基化的降低在TSS和TES中都很明显,在基因体中尤为突出。深入分析发现,初级精母细胞中DNA甲基化的下降发生在非翻译区(UTR)、转录本和RNA重复序列中;特别地,DNA甲基化在长末端重复序列(LTRs)和非LTRs(包括LINE和SINE)中也有所下降,这表明初级精母细胞中存在全基因组去甲基化现象。
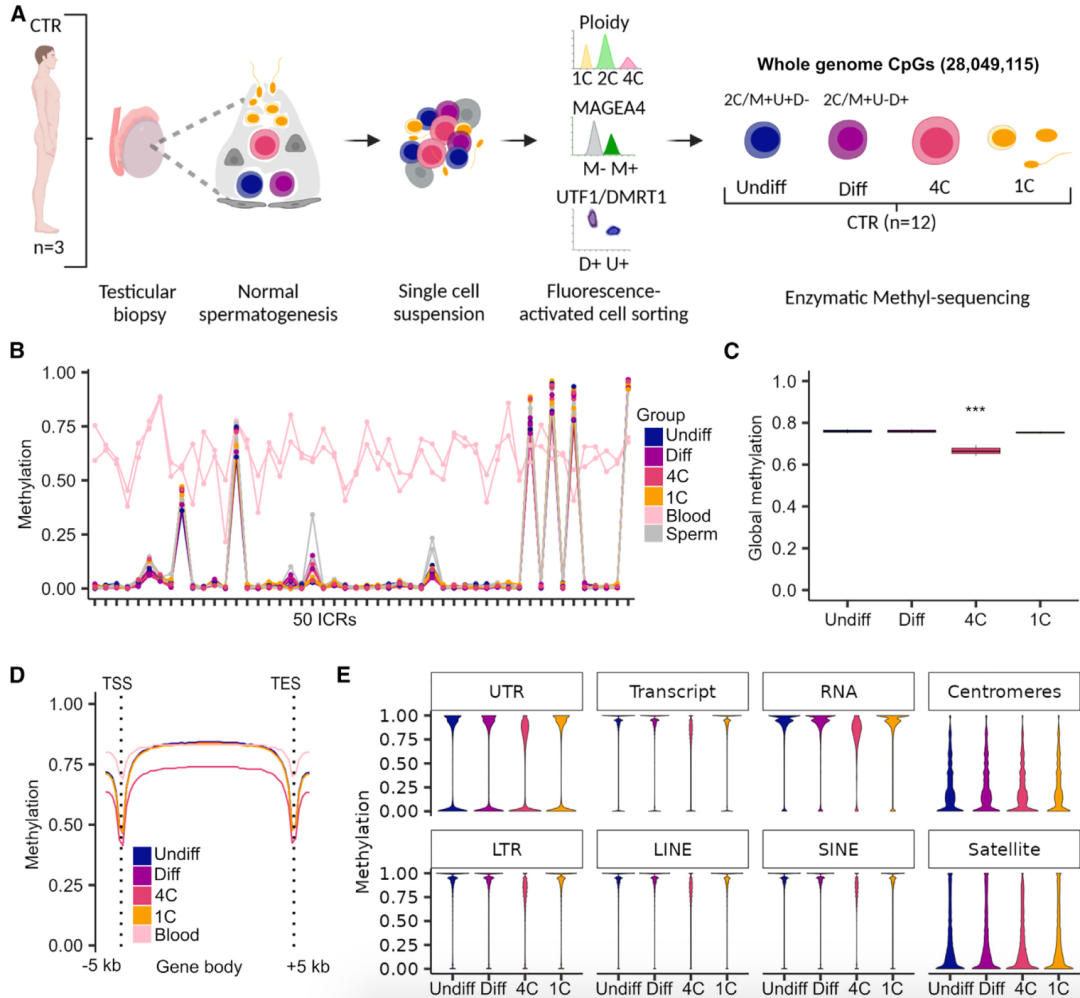
图1. 初级精母细胞表现出较低的全基因组DNA甲基化水平
DMRs在SINE重复位点富集
为确定在精子发生过程中DNA甲基化改变的区域,研究团队比较了不同生殖细胞类型的甲基化数据。经严格的过滤,共鉴定出16,000个差异甲基化区域(DMRs),并发现未分化精原细胞和分化精原细胞之间的DMRs最少,表明其甲基组具有高度相似性;在精原细胞和初级精母细胞之间检测到最多的DMRs。DMRs与基因或启动子的交叉分析结果显示,53%-70% DMRs与基因相关,3%-9% DMRs与启动子相关。
接下来,研究团队分析了DMRs的CpG富集度和长度,发现初级精母细胞DMRs中CpGs含量最高(平均14个)、长度最长(平均975 bp)。研究团队还探讨了DMRs是否对某些基因组特征显著富集,发现未分化精原细胞与精母细胞/精子的DMRs在CpG岛、3′UTR和外显子处显著富集,表明这些区域的DNA甲基化变化可能是精子发生过程的特征。有趣的是,不同生殖细胞类型之间的所有DMRs仅在SINEs区域显著富集,这表明在精子发生过程中SINEs的DNA甲基化变化发挥重要作用;LINEs重复区域的DNA甲基化优先被保留。
为揭示DNA甲基化变化在不同生殖细胞类型生物过程中的潜在调节作用,研究团队调查了已识别DMRs中具有调控区域的附近基因。结果显示,所有DMRs与29-1,685个基因的潜在调节区域重叠。GO术语分析表明,在精子发生过程中DNA甲基化变化与特定的细胞功能相关联。
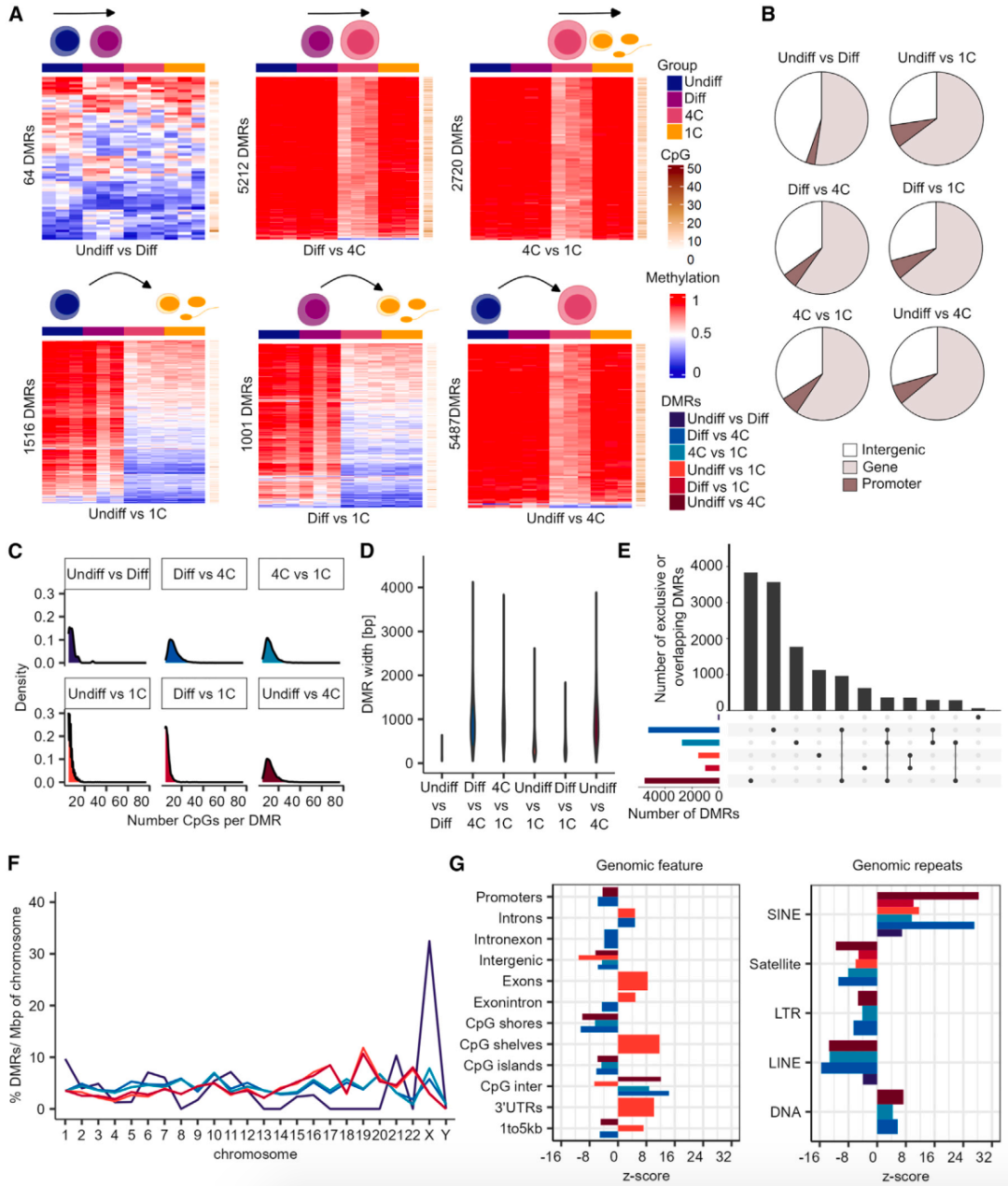
图2. 精子发生过程中的DMRs在SINE重复位点富集
低甲基化区域在转录因子结合位点富集
为研究精子发生过程中转录因子(TF)结合位点的DNA甲基化是否发生变化,研究团队分析了DMRs中TF结合基序的富集情况。结果显示,在精原细胞(未分化和正在分化)与精母细胞/精子之间比较的DMRs中,TF识别基序更丰富,包括DMRT1、DMRT6/DMRTB1和SOX6等。
上述基序存在差异甲基化,为此研究团队在已发表的scRNA-seq数据集中分析了其在精子发生过程中的表达。结果显示,DMRT1、DMRTB1和SOX6分别在精原细胞、精母细胞和精细胞的分化中存在特异性表达,这表明所鉴定基序的DNA甲基化变化是精子发生的一个特征。特别地,与未分化的精原细胞相比,精母细胞/精子的甲基组具有最多的低甲基化DMRs,并且富含TF结合位点。
为研究未分化精原细胞与精母细胞/精子的低甲基化DMRs是否标记了精子中的调控活性区域,研究团队分析了低甲基化DMRs与精子中保留的组蛋白(GSE40195)标记的重叠。结果显示,约17%的DMRs存在重叠,其中,精母细胞/精子中10%的低甲基化DMR与活性组蛋白标记H3K36me3重叠。

图3. 精细胞/精子中的低甲基化区域在转录因子结合位点富集
精子发生异常的甲基化改变
前期研究发现,在不育男性的精子中存在异常的DNA甲基化,特别是在印迹基因中。为将DNA甲基化变化与男性不育症联系起来,研究团队从被诊断为隐精子症(CZ)的男性(2名)睾丸组织中了分离生殖细胞,包括未分化精原细胞、分化精原细胞和主要精子细胞,并进行甲基化分析。
研究团队检查了CZ受试者与CTR(精子发生正常)之间是否存在DMRs。结果显示,在CTR组(精子发生正常)和CZ样本的未分化精原细胞、分化精原细胞和初级精母细胞中发现了107、144和152个DMRs;在所有比较中几乎没有重叠的DMRs。DMRs总体上富集于21号染色体和性染色体X,并显著富集于基因间区和反转录转座子,表明CZ生殖细胞中DNA甲基化模式的改变主要影响这些区域。
此外,研究团队还在CZ和CTR样本中分析了进化较年轻TEs(SVA D/F、L1Hs和L1PA2-5)和较老的TEs(HERVH-int和L1M7)的DNA甲基化变化。结果显示,CZ精原细胞(未分化和正在分化)和初级精母细胞在SVA D/F TEs中甲基化降低;SVA A TEs在所有CZ和CTR样本中都是高甲基化的。
在CZ样本的大量生殖细胞中,DNA甲基化的变化与功能相关的基因组区域有关。分析显示,DMRs分别与未分化精原细胞、分化精原细胞和初级精母细胞中29、39和38个基因的推测调控区域相关,这些区域显著富集了参与分化过程的基因。基因表达分析显示,25%-33%DMRs相关基因与精原细胞(PCDH11X、EDA等)、精母细胞(HSPBAP1、FOXK1等)或精子(CCDC200、AL589935.1等)特异性表达的转录本基因重叠。上述结果表明,CZ生殖细胞的DNA甲基化发生了变化,特别是在进化较年轻的SAV家族TEs和精子发生基因上。
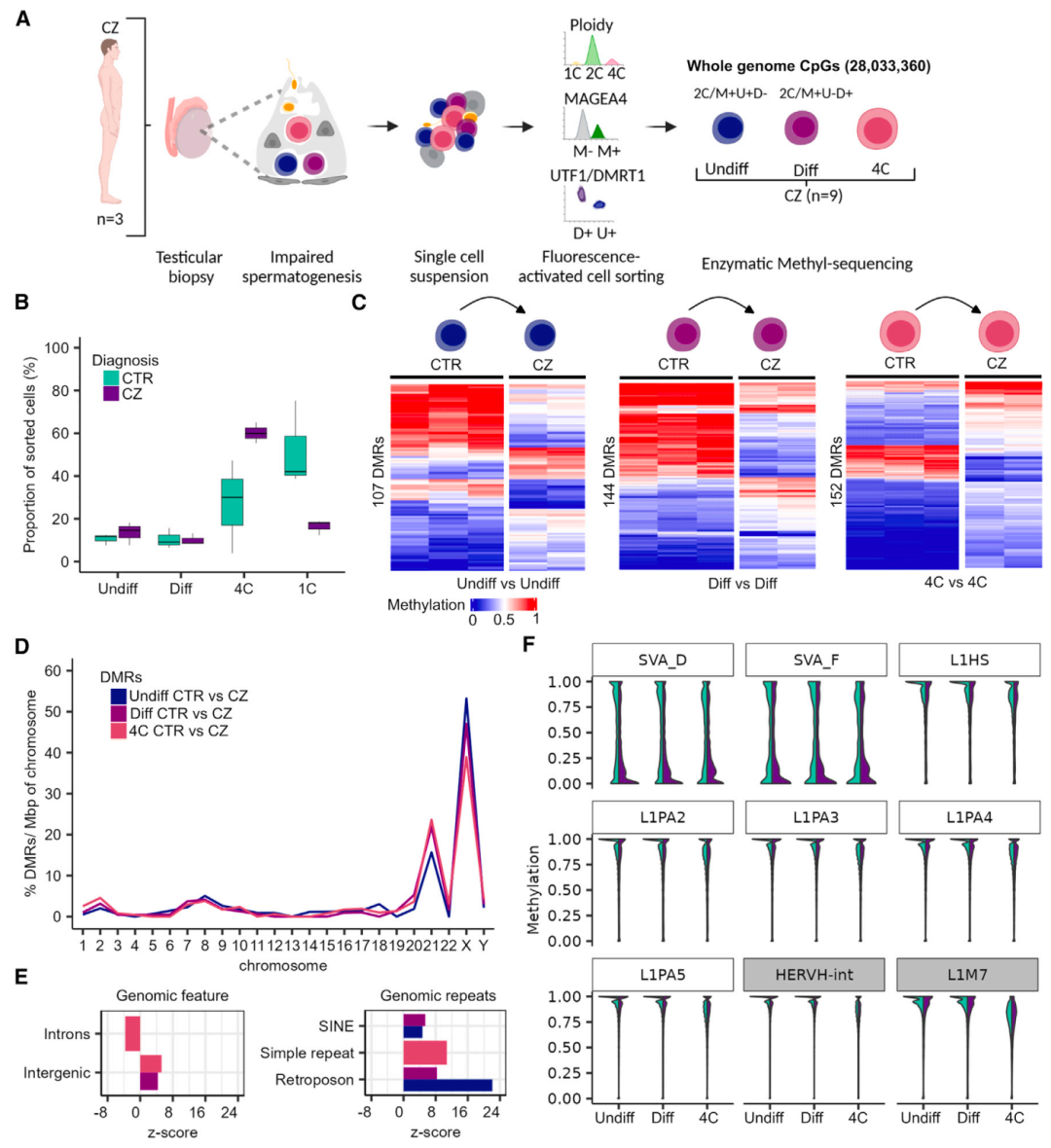
图4. 精子发生异常的甲基化改变
结 语
综上所述,研究团队描述了人类精子发生过程中全基因组DNA甲基化的变化及其在精子发生紊乱时的变化,发现精母细胞/精子中的低甲基化区域富含DMRT和SOX家族成员以及精子特异性基因转录因子的结合位点;在精子发生紊乱的情况下,生殖细胞表现出相当大的DNA甲基化变化。总之,该研究提供了人类精子发生过程中全基因组DNA甲基化变化的详细数据,突出了DNA甲基化(特别是TE)在这一过程中的关键作用,并指出TE甲基化改变在人类男性不育的病因学中的潜在作用。
参考原文:
Siebert-Kuss et al., Genome-wide DNA methylation changes in human spermatogenesis, The American Journal of Human Genetics (2024), https://doi.org/10.1016/j.ajhg.2024.04.017.

